
18β甘草次酸对17β羟基类固醇脱氢酶Ⅲ表达的抑制及诱导前列腺癌细胞LNCaP凋亡的研究
2012-09-07 09:14李泽良孙丹杨宇翀许晓龙孔垂泽
中国医科大学学报 2012年10期
李泽良,孙丹,杨宇翀,许晓龙,孔垂泽
(中国医科大学附属第一医院泌尿外科,沈阳 110001)
前列腺癌(prostate cancer,PCa)是男性恶性肿瘤致死的第二大疾病,目前对该病的最佳治疗方案和最有效的治疗药物尚未达成共识[1,2]。抑制雄性激素的生物合成及在其同源受体的活性在PCa的治疗中具有决定性意义。前列腺内部分有活性的雄性激素来源于由肾上腺分泌的活性甾体激素的前体脱氢表雄酮(dehydroepiandrosterone,DHEA)和雄烯二酮(androstenedione,ADT)[3,4]。17β 羟基类固醇脱氢酶Ⅲ(17βhydroxysteroiddehydrogenaseⅢ,17βHSD3)是介导最后一步由无活性前体向有活性激素转化的酶,是 17βHSD 家族成员之一[5~7],该家族对 PCa、乳腺癌等激素依赖性疾病有直接作用。因此,针对17βHSD3的小分子靶向抑制剂的研究已成为PCa研究的新热点。研究发现,18β甘草次酸(18β-glycyrrhetinic acid,18β-GA) 对 17βHSD3 具有抑制作用,但其作用机制尚不明确[8]。本研究拟寻找18βGA作用于PCa细胞的信号传导通路,明确其抑制17βHSD3的表达及PCa细胞生长的作用机制。
1 材料与方法
1.1 材料
雄性激素依靠性PCa细胞株LNCaP购自中国科学院上海生命科学研究院细胞资源中心;17βHSD3-pcDNA3稳定转染过表达HEK293细胞系受赠于美国新泽西州普林斯顿药物研究机构的Matthew V.Lorenzi教授。18β-GA 购自 Sigma-Aldrich公司;MTT购自Applygen Technologies公司;抗P-eIF2α及抗活化转录因子4(activating transcription factor 4,ATF4)抗体均购自 Santa Cruz Biotechnology公司;用于17βHSD3mRNA Real-time qPCR检测及CypA的引物均由Sigma-Aldrich公司设计并合成;eIF2α对应的siRNA及scramble RNA、脂质体类转染试剂 Lipofectamine LTX购自Invitrogen公司;SYBR Green 试剂盒购自 Toyobo 公司;Trizol、cDNA合成试剂盒购自TaKaRa公司。
1.2 方法
1.2.1 细胞培养:LNCaP细胞及17βHSD3-pcDNA3稳定转染细胞培养于含10%FBS及1%青霉素的DMEM培养基中,37℃、5%CO2孵箱中培养。17βHSD3-pcDNA3稳定转染细胞系培养时另加入500 ng/mL的G418,以筛除非目标细胞。
1.2.2 MTT检测:将LNCaP细胞加入至96孔板中(5 000/孔),37℃,5%CO2孵箱中培养 4 h。细胞贴壁后,更换培养基,将含有不同浓度18β-GA(终浓度:0,4,8,12,16 μmol/L) 的培养基 100 μL 加入孔内。继续培养 48 h,再次更换培养基(100 μL/孔),加入MTT溶液20 μL/孔,继续培养4 h。酶联免疫检测仪检测450 nm处各孔的吸光值。每组设4个复孔,重复3次。各组吸光值用±s表示。
1.2.3 Western blot:提取各组细胞全蛋白,取 20 μg蛋白行SDS-聚丙烯酰胺凝胶电泳。电转移至硝酸纤维素膜PVDF。用含1%脱脂奶粉的PBST封闭1 h,一抗溶液(用0.1%BSA稀释2 000倍)室温下振荡孵育1 h,加入辣根过氧化物酶标记的二抗溶液(用PBST稀释10 000倍),室温下振荡孵育1 h。碱性磷酸酶法显色。β-actin作内参照。实验重复3次。
1.2.4 RNA的提取及Real-time qPCR:细胞以1×105/孔传代于12孔板中培养4 h,细胞贴壁后,更换含 18β-GA(终浓度 8 μmol/L)的培养基,继续孵育48 h后回收。对照组以等体积的DMSO作为通用对照。PBS洗3次,用Trizol及氯仿抽提细胞内总RNA;紫外分光光度计测量浓度,每个样本使用1 μg的RNA进入下一步实验;用TaKaRa的反转录试剂盒合成cDNA;使用SYBR Green试剂盒进行Real-time qPCR反应,PCR的反应条件为:95℃10 min,反应 40 个下述循环:95 ℃ 20 s,54 ℃ 30 s,72℃30 s。CypA作内参照。
1.2.5 细胞转染:LNCaP细胞传代于12孔板中(1×105/孔),4 h细胞贴壁后,更换不含抗生素的培养基。以800 ng质粒稀释于200 μL OPTI-MEM中,加入质脂体类转染试剂Lipofectamine LTX 4 μL,静置30 min,缓慢加入培养基中,4 h后,更换不含抗生素的新鲜培养基,继续培养20 h。
1.3 统计学分析
2 结果
2.1 18β-GA可显著抑制LNCaP细胞的增殖
MTT法检测结果如图1所示:终浓度8,12,16 μmol/L的18β-GA作用48 h时,均能显著抑制LNCaP细胞的增殖(P<0.05);而浓度为4 μmol/L时,对细胞增殖的抑制作用不明显。故选择终浓度8 μmol/L的18β-GA进行后续实验。另外,如图2所示:终浓度8 μmol/L的18β-GA作用24 h时对细胞增殖的抑制作用不明显,但在48 h及72 h时,则能够显著抑制LNCaP细胞增殖(P<0.05)。因此,18β-GA对LNCaP细胞增殖的抑制作用具有剂量和时间依赖性。

2.2 18β-GA对LNCaP细胞中17βHSD3mRNA表达的抑制
Real-time qPCR结果如图3所示:8 μmol/L的18β-GA作用LNCaP细胞48 h后,细胞内17βHSD3 mRNA的表达水平较DMSO对照组显著降低(P<0.05)。

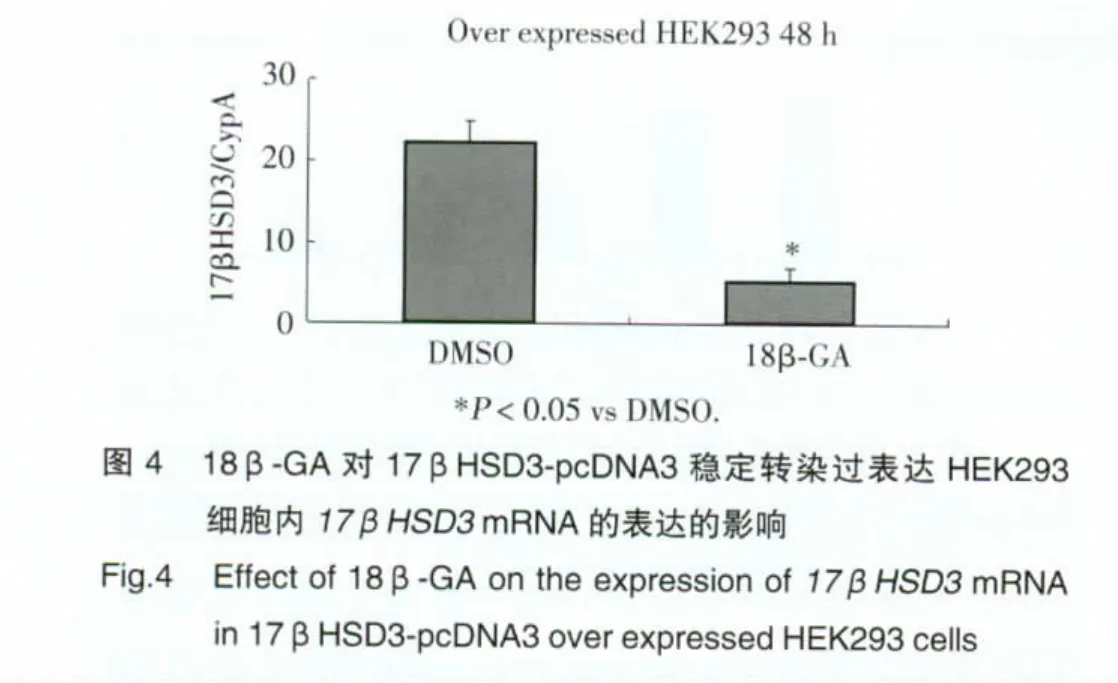
2.3 18β-GA对 17βHSD3-pcDNA3稳定转染过表达HEK293细胞系中17βHSD3mRNA表达的抑制
Real-time qPCR检测结果如图4显示:8 μmol/L 18β-GA作用17βHSD3-pcDNA3稳定转染过表达HEK293细胞48 h后,细胞内17βHSD3mRNA的表达水平显著降低(P<0.05)。
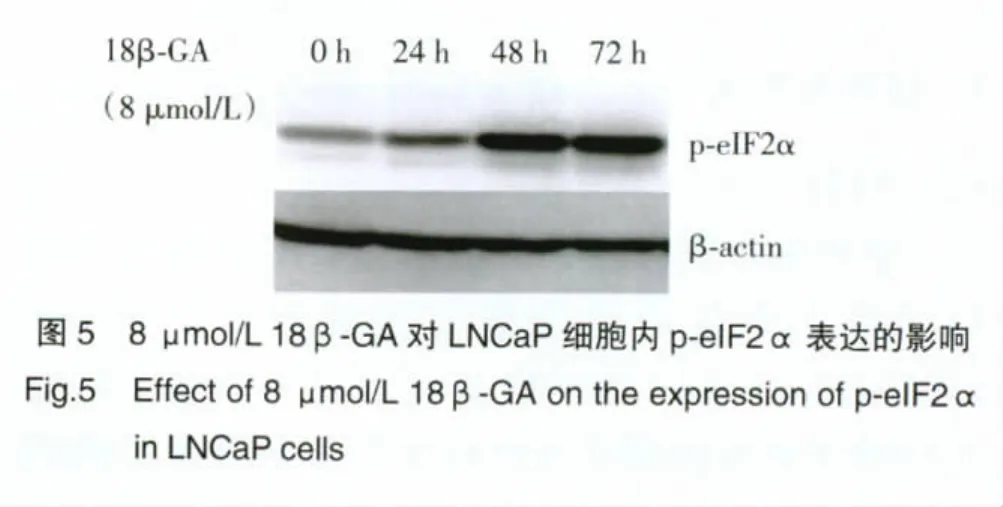
2.4 18β-GA通过使eIF2α磷酸化抑制17βHSD3 mRNA的转录
Western blot结果如图5所示:18β-GA的作用下,p-eIF2α的表达于24 h开始出现增强,48 h达到最大值。
2.5 18β-GA选择性增加ATF4的表达
Western blot结果如图 6所示:18β-GA激活eIF2α磷酸化后,降低了细胞内整体RNA转录开始的频率,但却选择性的增加了ATF4mRNA的转录,进而通过ATF4的靶向基因诱导细胞凋亡。

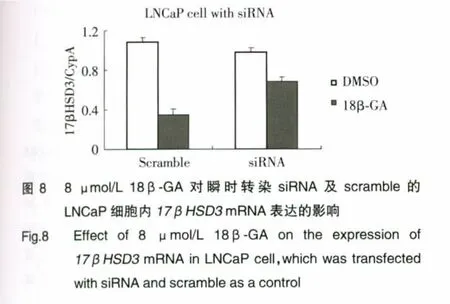
2.6 siRNA敲除eIF2α基因可明显减弱18β-GA对17βHSD3mRNA转录的抑制作用
我们首先确认了siRNA的基因干扰效率,结果如图7所示:当通过瞬时转染siRNA敲除eIF2α基因后,即使在18β-GA作用下,p-eIF2α蛋白的表达亦明显减少。随后我们检测了18β-GA对17βHSD3 mRNA转录水平的抑制效果,结果如图8所示:sieIF2α组18β-GA抑制17βHSD3mRNA的效果明显减弱(P<0.05)。

3 讨论
PCa是一种雄性激素相关性疾病。目前针对PCa激素水平的调控主要是通过抑制雄性激素的生物合成及其在同源受体的活性达到治疗目的[9,10]。但有研究表明,应用不同的内分泌治疗药物抑制其同源受体活性的方法不可避免的引起了骨质疏松、抑郁症等严重的不良反应[11]。因此,抑制雄性激素的生物合成在PCa治疗中相对更为安全有效。
外周组织中,有活性的雄性激素可以通过外周血中含量很高的无活性的激素前体的转化在靶器官中直接合成,17βHSDs家族是催化这一系列生化反应的关键酶。近年来针对17βHSD3的小分子靶向抑制剂的筛选成为研究热点,而其作用机制尚不明确。本研究探讨了17βHSD3的小分子靶向抑制剂18β-GA对PCa细胞内质网应激(endoplasmic reticulum stress,ER stress)通路的作用机制。结果显示,18β-GA能显著抑制PCa细胞的增殖,并诱导了PCa细胞的凋亡。同时,18β-GA还介导了eIF2α磷酸化水平的增加,降低了包括17βHSD3mRNA在内的细胞整体转录水平。当eIF2α被siRNA干扰后,18β-GA对17βHSD3mRNA转录的抑制作用也明显减弱。提示18β-GA对17βHSD3mRNA转录的抑制至少部分是通过eIF2α磷酸化这条通路介导的。磷酸化的eIF2α可以选择性增加包括ATF4在内的个别转录因子的表达[12],而ATF4可与CHOP的启动子上的AARE1顺式作用元件结合,从而诱导细胞凋亡[13]。因此,我们推测18β-GA诱导eIF2α磷酸化后,选择性地增加了ATF4的表达,并通过CHOP介导了PCa细胞的凋亡。本研究结果发现,18β-GA主要是通过ER stress通路同时实现了对癌细胞生长的抑制及对17βHSD3蛋白表达的调控。
本研究还发现,当eIF2α被siRNA干扰后,并未完全阻断18β-GA对17βHSD3mRNA的作用,所以我们推测18β-GA可能还存在其他的路径诱导细胞凋亡及调控酶水平的表达。今后,我们会继续探讨18β-GA可能作用的信号通路,同时寻找更多的17βHSD3靶向小分子抑制剂。
[1]Harada K,Kubo H,Abe J,et al.Discovery of potent and orally bioavailable 17β-hydroxysteroid dehydrogenase type 3 inhibitors[J].Bioorg Med Chem,2012,20(10):3242-3254.
[2]毕泗成,徐秀红,邵强,等.米非司酮对雄激素非依赖性前列腺癌LNCaPC4-2和PC3细胞的作用机制[J].首都医科大学学报,2004,25(2):250-252.
[3]EbelingP,KoivistoVA.Physiologicalimportanceofdehydroepiandrosterone[J].Lancet,1994,343(8911):1479-1481.
[4]Adams JB.Control of secretion and the function of C19-delta 5-steroids of the human adrenal gland[J].Mol Cell Endocrinol,1985,41(1):1-17.
[5]Mindnich R,Moller G,Adamski J.The role of 17 beta-hydroxysteroid dehydrogenases[J].Mol Cell Endocrinol,2004,218(1-2):7-20.
[6]Adamski J,Jakob FJ.A guide to 17beta-hydroxysteroid dehydrogenases[J].Mol Cell Endocrinol,2001,171(1-2):1-4.
[7]Mindnich R,Hrabe de Angelis M,Adamski J.Functional genome analysis indicates loss of 17beta-hydroxysteroid dehydrogenase type 2 enzyme in the zebrafish[J].J Steroid Biochem Mol Biol,2007,103(1):35-43.
[8]Spires TE,Fink BE,Kick EK,et al.Identification of novel functional inhibitors of 17 beta-hydroxysteroid dehydrogenase typeⅢ(17beta-HSD3)[J].Prostate,2005,65(2):159-170.
[9]Scher HI,Buchanan G,Gerald W,et al.Targeting the androgen receptor:improving outcomes for castration-resistant prostate cancer[J].Endocr Relat Cancer,2004,11(3):459-476.
[10]Roach M.Hormonal therapy and radiotherapy for localized prostate cancer:who,where and how long?[J].J Urol,2003,170 (6 Pt 2):S35-S40.
[11]Kruchten P,Werth R,Bey E,et al.Selective inhibition of 17betahydroxysteroid dehydrogenase type 1(17betaHSD1)reduces estrogen responsive cell growth of T47-D breast cancer cells[J].J Steroid Biochem Mol Biol,2009,114(3-5):200-206.
[12]Liu XA,Song J,Jiang Q,et al.Expression of the hyperphosphorylated tau attenuates ER stress-induced apoptosis with upregulation of unfolded protein response[J].Apoptosis,2012,17(10):1039-1049.
[13]Zong ZH,Du ZX,Li N,et al.Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells[J].Biochim Biophys Acta,2012,1823(8):1395-1404.
猜你喜欢
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
波谱学杂志(2022年1期)2022-03-15
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
中华建设(2019年7期)2019-08-27
天津医科大学学报(2019年6期)2019-08-13
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
