
贵州少数民族遗传性痉挛性截瘫3家系临床特点分析
2012-01-26 02:55李小丽
中风与神经疾病杂志 2012年8期
李小丽, 楚 兰, 徐 坚
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)又称Strumpell-Lorrain病,典型表现是以双下肢进行性肌张力增高、肌无力和剪刀步态为特征的综合征。由于本病具有高度遗传异质性[1],临床表现及遗传方式差异性较大。遗传方式分为常染色体显性、常染色体隐性、X连锁隐形遗传;临床表现分为单纯型和复杂型。复杂型HSP的临床特点表现更是多样性,除了痉挛性截瘫外,常合并脊髓外损害,如肌肉萎缩、精神发育迟滞、共济失调、多发性神经病、视神经萎缩、视网膜色素变性、耳聋、椎体外系等症状。故临床诊断、分型困难,基因诊断为“金标准”。
本文对我们发现的贵州地区少数民族(布衣、苗、彝)遗传性痉挛性截瘫3家系的患者及部分成员的临床特点进行分析。
1 临床资料
3个家系分别来自贵州省大方县、织金县、六盘水市,经家系调查发现3家系的遗传方式均符合常染色体显性遗传(见图1、图2、图3)。其中参与家系调查的HSP患者共14例:家系1(布衣族)共传递了3代,4例患者,2例已死亡;家系2(苗族)共传递了5代,包括14例患者,其中3例已死亡,现证者中3例(Ⅲ14、Ⅲ15、Ⅲ20)未参与本次家系调查;家系3(彝族)传递了5代,患者12例,8例已死亡。由神经内科2名专科医师对家系内所有成员进行体格检查,各成员的临床表现及病史如下。
1.1 家系1 布依族,连续3代发病,共4位患者。目前存活的患者只有2例,均为男性。因时间久远,已故的2位患者临床资料欠缺。Ⅲ1为先症者,41岁,男性,病程1年,起病表现为双下肢无力,逐渐出现行走不稳,言语含混,语速减慢。神经系统查体:双眼水平眼震,双侧膝反射活跃,昂伯征(+),双手轮替试验(+),步基增宽,一字步不能完成,双下肢肌力下降、肌张力(⧻),腱反射活跃,双侧病理征(+)。曾在当地医院行头部、胸腰段脊髓MRI等检查,未见明显异常。Ⅲ5,36岁,男性,病程10年,起病表现为行走不稳,左右摇晃,不能控制身体平衡,似喝醉酒样,多次住院治疗,未明确诊断。2008年在当地医院行头部MRI检查提示:轻度脑萎缩。已完全卧床2年,生活不能自理,双手不能持物,言语含混,轻度吞咽困难。神经系统查体:四肢肌张力明显增高,肌力3级左右,双侧膑阵挛,双侧Babinski征(+),未见明显肌萎缩。家系内其他成员除个别出现双侧膝反射对称性活跃外,未见其他阳性发现。已故的两位患者为现症者的父亲及祖父,根据家族成员回忆描述,他们两位生前均在青壮年起病,开始表现为行走不稳,双下肢无力,后逐渐言语不清,肢体发僵,最后卧床不起,具体死因不明。该家系患者成员均有“行走不稳”的临床表现,有的甚至是首发症状,现症患者神经系统查体均有共济运动障碍,提示该家系不是典型单纯性HSP。根据明显的家族史,结合患者锥体束征,诊断为复杂型HSP。不难看出,该家系前3代每代均出现患者,第4代共8位成员,目前尚未出现病例,是遗传学上的自然选择还是症状尚未显露?有待进一步跟踪随访。
1.2 家系2 第一代患者为苗族,连续5代发病,男女均罹患。遗憾的是,由于家系内一部分成员长期在外地定居,通讯联系不上,本次家系调查不能参加,他们的情况只能由其他家庭成员描述。V9(先证者)为12岁,男性,2岁起即无明显诱因出现双下肢僵硬、迈步费力,易跌倒,跑步时更加明显,病情逐渐加重,神经系统查体:双下肢腱反射活跃,肌张力呈折刀样增高,双侧病理征阳性。双上肢肌力、肌张力正常,住院期间行胸髓MRI检查未见明显异常,肌电图检查提示神经源性损害。给予肌松剂(盐酸乙派立松)治疗,双下肢僵硬、行走无力有所缓解。但随访半年仍未恢复正常。该患者与本家系体检发现的另2位患者(Ⅳ37、Ⅴ10)均在童年发病,与家系其他成员比较发病较早,体现了遗传早现现象。Ⅱ5为67岁,男性,27岁起病,40余年病史,因经济条件差,一直未予治疗。该患者主要表现为双下肢无力,行走不稳,近2年出现饮水呛咳,吞咽困难,伴双下肢麻木。神经系统查体:智能下降,四肢腱反射活跃,双侧踝阵挛,双侧Babinski征(+),剪刀步态,双下肢痛觉减退,振动觉、运动觉未见异常。Ⅲ1,男性,61岁,病史3年,因双下肢无力,行走不稳起病,查体:双下肢肌力4级,双侧腱反射活跃,双侧踝阵挛,但双侧病理征(±),双侧膝关节以下痛觉及关节位置觉减退。Ⅲ7为56岁,女性,48岁起病,以双足底烧灼感为首发症状,双足尖抬高困难,行走拖步,易摔倒,伴眩晕。神经系统查体:四肢肌张力呈铅管样增高,下肢重于上肢,双侧Babinski征(+),共济运动障碍,该患者伴明显锥体外系症状,表现为复杂型HSP。Ⅲ16,35岁发病,21年病程,首发症状为双上肢麻木,逐渐出现四肢僵硬。查体:肌张力明显增高,下肢重于上肢,双侧Babinski征(+)。Ⅳ12、Ⅳ37、Ⅴ10无自觉症状,体检时发现双下肢腱反射亢进,踝阵挛,双侧Babinski征(+)。Ⅲ2为61岁,男性,3年病程,主诉四肢无力,行走拖步。神经系统查体:双上肢肌张力呈齿轮样增高,双下肢肌张力呈铅管样增高,四肢肌力5级,四肢腱反射正常,双侧Babinski征(-),行走时连带动作消失。诊断为“帕金森病”,给予“美多芭”治疗后症状缓解。Ⅲ10为53岁,男性,因右侧肢体无力、不自主抖动起病,后缓慢出现吞咽困难、饮水呛咳。神经系统查体:静止性震颤,面具脸,四肢肌张力呈齿轮样增高,四肢腱反射正常,双侧Babinski征(-),亦诊断为“帕金森病”,给予“美多芭”治疗后症状缓解。Ⅲ2、Ⅲ10为家系内同一代患者,以锥体外系为首发症状,未发现明显锥体束征,且对多巴胺制剂治疗有效,目前诊断为“帕金森病”是适当的。但该2例患者是HSP复杂型的早期表现还是单纯帕金森病,以及后期病情的发转转归,有赖于我们对病情的进一步跟踪随访。总之,该家系内参与家系调查的10例生存患者临床表现差异性大,6例单纯型,2例复杂型,2例表现为PD症状。
1.3 家系3 本家系较大,彝族,共传递了5代,男女均有罹患。结果为12例患者,8例已死亡,自愿参与该家系调查的共27人(家系图谱见图1)。先证者Ⅳ1,36岁,男性,3年前因行走不稳起病,伴下肢无力,近一年出现上肢无力,伴言语含混不清、饮水呛咳、双小腿酸胀。神经系统查体:双眼水平眼震,一字步不能完成,双侧膝反射活跃,双侧病理征阳性。感觉系统未见明显异常。该患者已生育3个男孩,最小的才6个月,目前3个男孩均未出现临床症状。Ⅳ3为46岁,男性,无自觉症状,查体发现四肢轻微静止性震颤,右上肢肌张力齿轮样增高,考虑为早期“帕金森病”。然而,与家系1中Ⅲ2、Ⅲ10帕金森表现一样,HSP家系中PD的高发现象,我们推断可能与HSP基因在黑质-纹状体区过度表达有关,值得我们进一步探究。Ⅳ27与V23、V24为父子关系,临床症状均较重。Ⅳ27为30岁,男性,病程8年,丧偶,表现为行走不稳,查体可见Ⅱ度水平眼震,膝反射(),可引出踝阵挛,双手指鼻不准,昂伯(-),双侧Babinski征(+)。V24为7岁,男童,查体可见Ⅱ度水平眼震,膝反射(⧻),未引出膑阵挛,双侧病理征阳性。V25为4岁,男童,拿勺吃饭时双手不自主大幅摆动,几乎不能将食物送入口,查体可见Ⅱ度水平眼震,双下肢肌张力折刀样增高,膝反射(),可引出踝阵挛,双侧病理征阳性,双侧指鼻不准,昂伯征(+)。该家系只有4例生存患者,其临床表现均为复杂型,以共济失调为主,且呈明显的遗传早现现象。
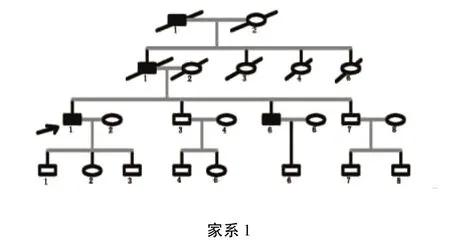
图1 布衣族HSP家系图谱
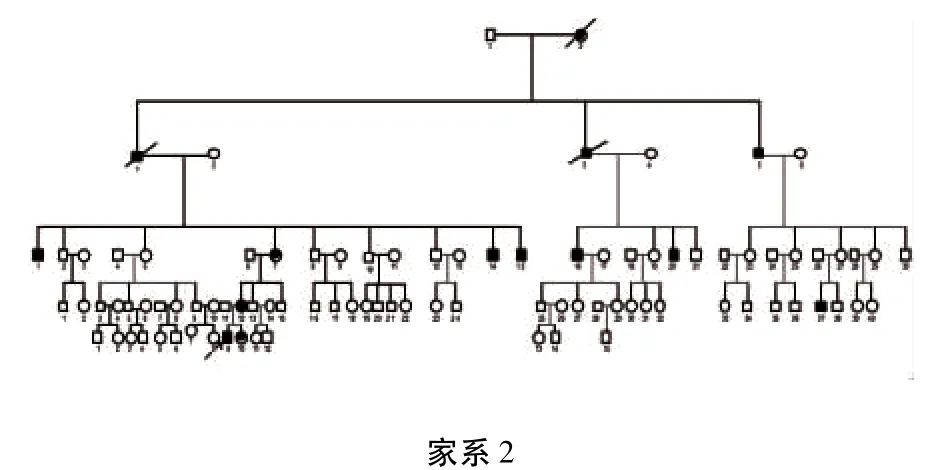
图2 苗族HSP家系图谱
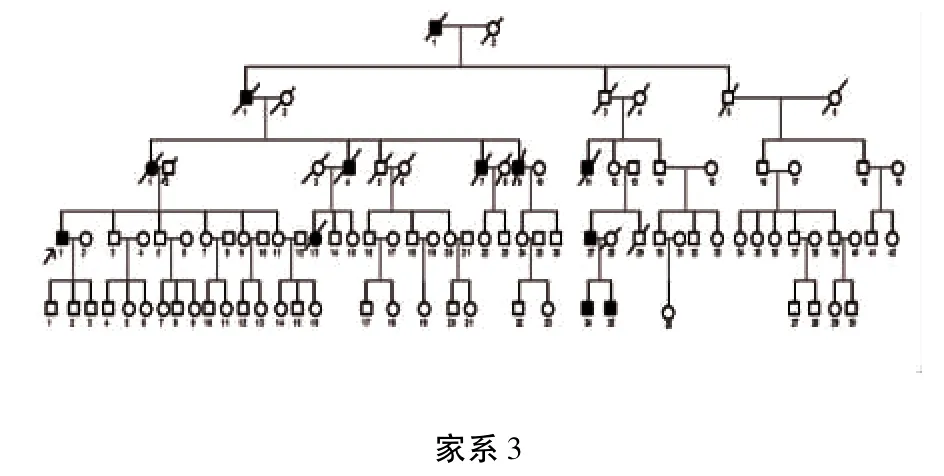
图3 彝族HSP家系图谱
2 结果
通过对上述3家系的临床特点归纳分析可发现,3个家系均为常染色体显性遗传;现证者男性明显多于女性(11∶3);复杂型比例较高,占71.4%(10/14),且最常见的合并症为共济失调(7/10);遗传早现现象明显(2/3);HSP家系中PD的发生率较高(2/3)。
3 讨论
HSP是一组具有明显临床和遗传异质性的神经变性疾病,以 AD遗传为主,AD-HSP外显率为95%[2]。从家系图可以看出,本研究3家系均符合AD遗传规律。根据AD的遗传特征,家系3Ⅱ3应为表现型,但该患者呈正常型,推测其原因:(1)由于AD-HSP外显率只有95%,该患者呈不完全显性;(2)患者死亡多年,年代久远,资料失真。家系2和家系3均呈现遗传早现现象,以前认为该病多发生在儿童或青少年,但从文献报道[3]及本组研究结果看,其发病年龄跨度较大,从婴儿期到老年患者均有发病,婴儿期诊断率低可能与症状不易发现有关。本组资料发病年龄最大58岁,最小2岁,18岁以前发病 5例,占 35.7%,18岁以后发病 9例,占64.3%。男性11例,女性3例,与文献报道的HSP患者男多于女一致[4],这种性别上的“女性保护”现象可能与生化代谢过程中性别参与的修饰因子等作用有关[5]。1981年Harding教授提出单纯型HSP诊断标准[6]:(1)10~50岁起病。(2)缓慢进展的双下肢痉挛性无力,肌张力增高,腱反射亢进,病理征阳性,剪刀步态等症状和体征。(3)大部分病例有阳性家族史。1983年Harding教授又将HSP分为单纯型和复杂型[7]。单纯型又称stumpellⅡ型,患者逐渐感双下肢僵硬,伸肌张力高,下肢腱反射亢进,病理反射阳性,剪刀步态,晚期可出现上肢锥体束征、四肢共济失调等体征。复杂型除具有单纯型的病理征以外,尚合并小脑性共济失调、震颤、多发性神经病、肌萎缩、癫痫、胼胝体发育不良、听力障碍、智能减退,可构成不同的综合征[8,9],约 50% 病例报道有脊髓小脑束受累[10]。本组患者中复杂型10例,占全部患者71.4%(10/14),这远高于外国文献报道[2]。
复杂型HSP国外最常见的合并症是肌萎缩[11],中国最常见的是痴呆[12]。本组最常见的是共济失调占70%(7/10),其次是眼球震颤50%(5/10),再次是感觉障碍占40%(4/10),仅Ⅱ5合并有痴呆,Ⅲ3合并肌萎缩。值得一提的是,2003年赵国华等对中国39个家系113例HSP患者临床资料分析发现,该组复杂型HSP患者高达78.8%,且最常见的合并症亦是共济失调(46.9%)[13],均与本组数据较吻合,很可能提示中国人复杂型HSP发生率高于外国人,且中国人复杂型HSP合并症中最常见的是共济失调而不是痴呆。然而,本组中3家系中有2家系出现发病年龄相对较早的PD患者,这种HSP家系中“PD聚集”现象未见有文献报道,值得进一步探究。
有研究显示,显性遗传性复合型HSP伴发痴呆患者的发病年龄较其他类型晚,智能减退多发生在疾病的晚期[14],这与Ⅱ5患者40岁发病、病程长达28年一致。HSP患者泌尿系症状国外报道的较多,但本组未见。本组共4例患者出现感觉障碍,占复杂型HSP患者的40%,高于文献报道。弓形足是HSP的常见体征,但本组无患者发生。
遗传性痉挛性截瘫和遗传性共济失调很多症状重叠,分型困难,从临床症状学上看,多数学者认为前者是后者的一个亚型[15],但从遗传学上看,二者突变位点和突变形式均不同,是两个独立遗传疾病。总之,本组HSP患者除表现双下肢痉挛性无力外,还表现有锥体外系症状、感觉障碍等。但是,并非所有的HSP患者均表现双下肢肌张力增高,本组有2例(家系1中的Ⅲ1、家系2中的Ⅴ10)患者肌张力正常,占14.3%(2/14),肌力亦无明显下降,其原因考虑为:(1)体检者主观误差;(2)该2例患者尚处于疾病早期,一些临床体征尚未显露。通过表1可以看出,本组患者中病程最长者28年,最短者1年,病程越长,下肢的剪刀步态越明显。
综上所述,贵州地区少数民族HSP临床特点与文献报道有较大差异。当然,需扩大样本研究。我们正在进一步搜集我国少数民族HSP家系及临床特点,建立数据库,并对患者基因突变进行分析,以期从分子水平寻找我国少数民族HSP遗传及临床特点。
[1]吴 江.神经病学[M].北京:人民卫生出版社,2011.358-360.
[2]McDermott CJ,White K,Bushby K,et al.Hereditary spastic paraparesis:a review of new developments[J].J Neurol Neurosurg Psychiatry,2000,69:150 -169.
[3]McDermott CJ,Burness CE,Kirby J,et al.Clinical features of hereditary spastic paraplegia due to spastin mutation[J].Neurology,2006,67(1):45-51.
[4]Starling A,Rocco P,Passos-Bueno MR,et al.Autosomal dominant(AD)pure spastic paraplegia(HSP)linked to locus SPG4 affects almost exclusively males in a large pedigree[J].Med Genet,2002,39:77.
[5]Orlacchio A,Kawarai T,Gaudiello F,et al.Clinical and genetic study of a large SPG4 Italian family [J].Mov Disord,2005,20:1055 -1059.
[6]Harding AE.Genetic aspects of autosomal dominant late onset cerebellar ataxia[J].Med Genet,1981,18:436 -441.
[7]Harding AE.Classification of the hereditary ataxias and paraplegias[J].Lancet,1983,1(8334):1151 -1155.
[8]Nielsen JE,Johnsen B,Koefoed P,et al.Hereditary spastic paraplegia with cerebellar ataxia:a complex phenotype associated with a new SPG4 gene mutation[J].Eur J Neurol,2004,11(12):817 - 824.
[9]Erichsen AK,Inderhaug E,Mattingsdal M,et al.Seven novel mutations and four exon deletions in a collection of Norwegian patients with SPG4 hereditary spastic paraplegia[J].Eur J Neurol,2007,14(7):809-814.
[10]陈 昕,赵国华,唐北沙.遗传性痉挛性截瘫的病理、遗传学、发病机制和临床的研究进展[J].临床神经病学杂志,2006,19(1):70-72.
[11]Harding AE.The hereditary ataxia and paraplegia.In principle and practice of medical genetics[M].ChurchillLivingstone,1990.383 -396.
[12]陈 嵘,龚黎民,梁秀龄.中国遗传性痉挛性截瘫的临床特点[J].中风与神经疾病杂志,1998,15:226.
[13]赵国华,唐北沙,罗巍等.遗传性痉挛性截瘫的临床和遗传特点[J].临床神经病学杂志,2003,16(1):31 -33.
[14]Lizcano-Gil LA,Garcia-Cruz D,del Pilar Bernal-Beltran M,et al.Association of late onset spastic paraparesis and dementia:probably an autosomal dominant form of complicated paraplegia[J].AM J Med Genet,1997,68:1 -6.
[15]蒋雨平,邬剑军.遗传性共济失调[J].中国临床神经病学,2011,19(5):515 -520.
猜你喜欢
基层中医药(2022年1期)2022-07-22
中华养生保健(2022年3期)2022-02-23
实用临床护理学杂志(电子版)(2020年27期)2020-07-15
云南中医中药杂志(2020年3期)2020-05-11
中西医结合心血管病杂志(电子版)(2020年3期)2020-04-21
健康大视野(2020年6期)2020-04-10
中成药(2019年12期)2020-01-04
制造技术与机床(2017年11期)2017-12-18
中国民族民间医药·下半月(2016年4期)2016-05-24
医学美学美容·中旬刊(2015年2期)2015-10-21
