
西司他丁钠的合成*
2011-12-17 09:41欧阳罗陈瑞业柏一慧李新生徐东成
浙江师范大学学报(自然科学版) 2011年3期
欧阳罗, 陈瑞业, 柏一慧, 李新生, 徐东成
(浙江师范大学化学与生命科学学院,浙江金华 321004)
西司他丁(Cilastatin)与亚胺培南的复方药——泰能,是临床上广为应用的碳青霉烯抗菌药[1].西司他丁本身不具有杀菌作用,但是它可以抑制亚胺培南在肾脏被脱氢二肽酶分解,从而提高亚胺培南的活性,因而其合成方法引起了人们的重视.西司他丁分子可以由3部分组成:(S)-(+)-2,2-二甲基环丙烷甲酰胺,庚酮酸衍生物和L-半胱氨酸,目前已有的文献都是从这3个片段着手合成西司他丁.Donald等[2]于1987年报道了一条通过合成消旋二甲基环丙烷甲酸,拆分后得到手性的二甲基环丙烷甲酸,进而得到手性的环丙烷甲酰胺,再与酮酸酯、半胱氨酸反应,最终获得西司他丁的路线,总收率为14.6%.其后,徐晓莉等[3]对其合成路线中的后处理过程进行了改进,硫醚化的产率由63%提高到79%;石晓华等[4]对其合成路线中的7-氯-2-氧代庚酸的合成进行了研究.到目前为止,在硫醚合成步骤中,所有的文献([2-4])都采用酸和半胱氨酸或者胱氨酸在强碱的作用下进行合成,由于2个原料和产品都是水溶性的,不但反应难于检测,而且产率低、产物复杂,对后面的提纯造成了很大的困难.另外,在西司他丁的合成中还有一个难题有待解决,即手性2,2-二甲基环丙烷甲酰胺的合成,其主要通过拆分消旋的2,2-二甲基环丙烷甲酸获得.文献报道的拆分剂有奎宁[2]、R-1-(3-甲氧基苯基)-乙 胺[5]、L-肉 碱 草 酸 盐[6]、L-薄 荷 醇[7]、(S)-扁桃酸甲酯[8]、L-肉碱盐酸盐[9]和顺式-2-苄胺基环己烷甲醇[10]等,这些拆分剂虽然能够拆分二甲基环丙烷甲酸,但是收率均在20% ~30%,相对较低.加上由酸合成酰氯再得到酰胺的步骤,使得酰胺的收率较低.近年来也有研究者[11-14]用金属络合物为催化剂,以不对称合成的方法合成二甲基环丙烷甲酸,但其所需的底物之一为重氮酯,很大程度上限制了其应用.鉴于合成中存在的问题,笔者尝试了用新的拆分剂和温和的反应条件对上述合成中的问题进行探讨,期望得到高光学纯度和合成收率的西司他丁钠.
1 实验部分
1.1 仪器与试剂
1H NMR,13C NMR在 Bruker AVANCE 400型核磁共振仪上测定,以CDCl3,D2O和氘代二甲基亚砜(DMSO-d6)为溶剂,四甲基硅烷(TMS)为内标;熔点由WRS-1B数字熔点仪测定,温度计未校正;旋光度由WZZ-2B自动旋光仪测定;非对映体过量(de)值用高效液相色谱法(HPLC)测定,色谱柱型号为Chiralcel OD-H,流动相为异丙醇/正己烷,溶剂使用前经无水处理.
1.2 西司他丁钠的合成
合成路线见图1和图2.
1.2.1 联萘酚单酯的合成
氮气氛围下,在100 mL圆底烧瓶中加入(R)-Ⅰ (2 g,6.98 mmol),Et3N(5.3 mL)和CH2Cl2(50 mL),搅拌,冷至-20℃,快速搅拌下缓慢滴加 2,2-二甲基环丙烷甲酰氯(0.93 g,7.01 mmol),滴加完毕后升温至室温,TLC(薄层色谱法)跟踪反应.反应完毕,有机层分别用水洗、饱和食盐水洗,有机层用无水Na2SO4干燥,减压蒸除溶剂,得到粗产物.粗产物经硅胶柱色谱纯化(洗脱剂V(乙酸乙酯)∶V(石油醚)=1∶8),得到(1R,2S)-Ⅱ 1.15 g,产率为 43.1%,和(1R,2R)-Ⅲ 1.3 g,产率为 48.7%.
产物(1R,2S)-Ⅱ:淡黄色固体,熔点为160.8 ~161.0 ℃;[α]20D为 +140.89(c为 5.15 mol/L,四氢呋喃(THF));de值为 97.7%;1H NMR(CDCl3,400 MHz)δ:0.73(s,3H),0.75(m,1H),0.95 ~ 0.96(m,1H),0.99(s,3H),1.34 ~1.37(m,1H),5.37(s,1H),7.05(m,1H),7.23~7.36(m,6H),7.39~ 7.41(m,1H),7.84 ~ 7.98(m,3H),8.05 ~ 8.08(m,1H);13C NMR(CDCl3,100 MHz)δ:18.1,22.8,24.4,26.4,26.7,114.3,118.3,122.2,123.3,123.5,124.8,125.8,126.2,126.6,127.4,127.9,128.3,129.1,130.2,130.7,132.2,133.7,133.7,148.3,151.8,172.5.
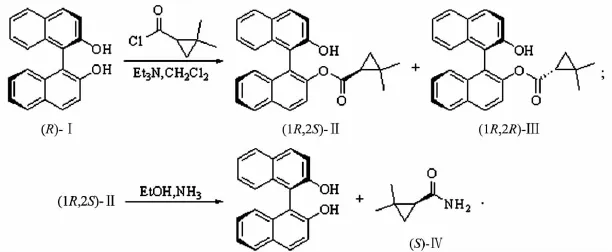
图1 手性二甲基环丙烷甲酰胺(Ⅳ)的合成
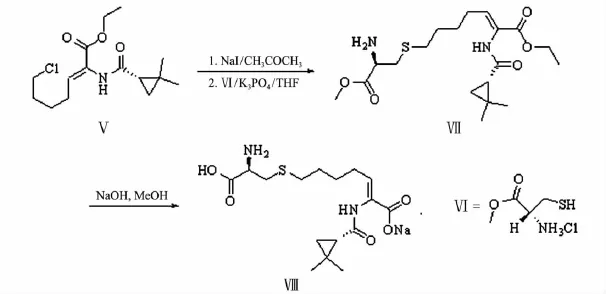
图2 西司他丁钠(Ⅷ)的合成
产物(1R,2R)-Ⅲ:淡黄色固体,熔点为116.5~124.1 ℃;[α]20D为 +74.36(c 为 5.14 mol/L,THF);de值为 94%.
1.2.2 手性2,2-二甲基环丙烷甲酰胺的合成
在 100 mL圆底烧瓶中,将(1R,2S)-Ⅱ(1.91 g,4.99 mmol)溶于无水乙醇(50 mL)中,在冰浴下通氨气至饱和,室温下密封反应,气相色谱跟踪检测至(1R,2S)-Ⅱ反应完全.减压蒸除溶剂,经硅胶柱色谱纯化(洗脱剂V(乙酸乙酯)∶V(石油醚)=1 ∶4),得到2,2-二甲基环丙烷甲酰胺(Ⅳ)0.56 g,产率99%,联萘酚回收率99%.
产物(S)-Ⅳ:白色固体,熔点为 139.0~140.5 ℃(文献值[2]:136.0 ~137.5 ℃);[α]20D为+97.2(c 为 1.5mol/L,CHCl3);1HNMR(DMSO-d6,400 MHz)δ:0.55 ~ 0.58(m,1H),0.78~ 0.81(m,1H),1.04 ~ 1.06(m,6H),1.34 ~1.38(m,1H),6.72(s,1H),7.39(m,1H);13C NMR(DMSO-d6,100 MHz)δ:18.9,19.6,20.7,27.3,27.8,173.2.
1.2.3 化合物Ⅴ的合成
按文献[15]合成 7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺基)-2-庚烯酸乙酯(Ⅴ).
1.2.4 西司他丁钠的合成
在100 mL圆底烧瓶中加入化合物Ⅴ(0.5 g,1.66 mmol),NaI(0.25 g,1.66 mmol)和丙酮(45 mL),加热回流12 h,反应完后过滤,滤液旋干,得7-碘-2-((S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯.
在氮气氛围下,在100 mL圆底烧瓶中加入上一步的碘代产物(0.22 g,0.56 mmol)和半胱氨酸甲酯盐酸盐 Ⅵ (0.14 g,0.84 mmol),K3PO4(0.47 g,2.23 mmol)和 THF(40 mL),在超声波中反应15 min,将反应装置置于已加热到80℃的油浴中回流反应11 h,反应完后冷却至室温,减压蒸除溶剂,将其溶于乙酸乙酯,有机相用水洗、饱和食盐水洗,无水Na2SO4干燥,粗产物经硅胶柱色谱纯化(洗脱剂V(乙酸乙酯)∶V(石油醚)=2 ∶1),得到产物Ⅶ(西司他丁酯),产率74.1%.
将产物Ⅶ(0.37 g,0.92 mmol)溶于甲醇(10 mL)中,加入氢氧化钠(0.15 g,3.7 mmol)和水(5 mL),室温搅拌3 h,反应完后加入 1 mol/L HCl溶液中和至中性,真空旋干,溶于甲醇,快速过滤,旋干得产物Ⅷ(西司他丁钠).
产物Ⅶ:淡黄色油状物;1H NMR(CDCl3,400 MHz)δ:0.77 ~ 0.80(m,1H),1.13 ~ 1.17(m,7H),1.27 ~1.30(m,3H),1.41 ~1.42(m,1H),1.54 ~1.59(m,4H),1.78(s,2H),2.12 ~ 2.14(m,2H),2.52(t,J=8 Hz,2H),2.73 ~ 2.76(m,1H),2.86 ~2.91(m,1H),3.61 ~3.64(m,1H),3.72(s,3H),4.18 ~ 4.23(m,2H),6.59(t,J=8 Hz,1H),7.08(s,1H);13C NMR(CDCl3,100 MHz)δ:14.2,18.7,20.6,22.5,27.1,27.3,28.7,29.3,32.2,37.2,52.2,54.1,61.4,125.3,136.7,164.9,169.7,174.5.
产物Ⅷ:淡黄色粉末;1H NMR(D2O,400 MHz)δ:0.75 ~ 0.77(m,1H),0.88 ~ 0.90(m,1H),0.95 ~1.00(s,6H),1.41 ~1.48(m,4H),1.55 ~ 1.57(m,1H),2.08 ~ 2.10(m,2H),2.46~ 2.49(m,2H),2.88 ~ 2.90(m,1H),2.96 ~2.97(m,1H),3.76 ~ 3.79(m,1H),6.42(t,J=8 Hz,1H).
2 结果与讨论
2.1 (S)-2,2-二甲基环丙烷甲酰胺的制备
(S)-2,2-二甲基环丙烷甲酰胺作为西司他丁合成的重要中间体,其产率的提高,对于合成西司他丁产率的增加有着至关重要的作用.2,2-二甲基环丙烷甲酰氯与(R)-联萘酚通过酯化高产率地得到 2-(2,2-二甲基环丙烷甲酸)-1,1'-联萘酚酯,柱色谱分离非对映体,需要的非对映体直接氨解,即可得到(S)-2,2-二甲基环丙烷甲酰胺.该方法得到的2,2-二甲基环丙烷甲酰胺光学纯度较高,氨解产率可达到99%,而且拆分试剂也可定量地回收.
2.2 西司他丁酯的合成
在西司他丁酯的合成中,以7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ)和半胱氨酸甲酯盐酸盐(Ⅵ)为反应物,考察了该反应较佳的碱性条件和较佳的溶剂,探讨了反应温度、碱、原料摩尔比等因素对反应产率的影响.
2.2.1 碱强度的影响
碱的强弱对该反应有着很重要的作用.以四氢呋喃(THF)为溶剂,原料Ⅴ,Ⅵ和碱的摩尔比为1∶1∶3时,考察了不同的碱对西司他丁酯产率的影响.实验结果证明,碱性过强或碱性过弱均对产物的生成有影响.从表1可以看出:以K3PO4作为反应用碱,西司他丁酯的产率较好,达到70.9%;强碱和弱碱都显著地降低产物的产率.对于碱性比 K3PO4稍微弱的碱,如 K2HPO4和KHCO3,产物的产率显著下降;对于碱性比K3PO4更弱的碱,如KH2PO4,则没有产物生成;而用碱性比K3PO4强的碱,如NaH,也基本没有产物生成.
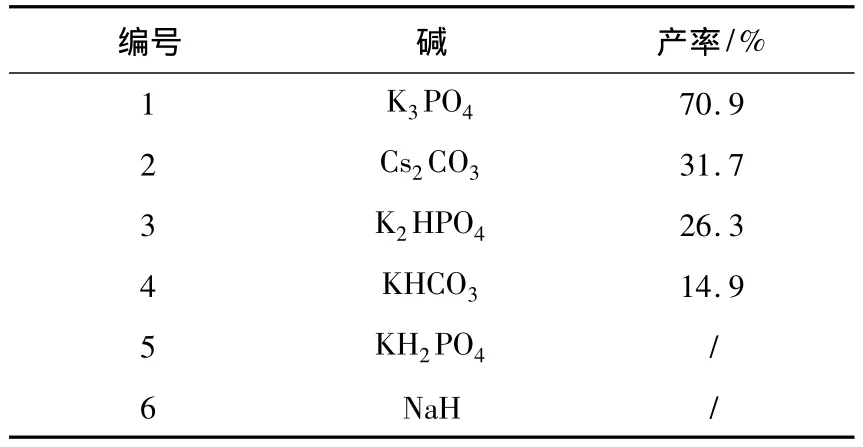
表1 碱强度对西司他丁酯产率的影响
2.2.2 溶剂的影响
溶剂的性质对反应有着重要的影响.在原料7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ),半胱氨酸甲酯盐酸盐(Ⅵ)和碱的摩尔比为1∶1∶3,K3PO4为碱时,考察了不同的溶剂对西司他丁酯产率的影响.从表2可以看出:以四氢呋喃(THF)为溶剂时产物的产率较高,达到70.9%;当溶剂为1,4-二氧杂环乙烷(dioxane)或甲苯(toluene)时,产物的产率降低;而当考虑用(C2H5)2O或CH2Cl2时,产率也降低较多;选择AcOEt或CHCl3为溶剂时,也给出了比THF稍低的产率.
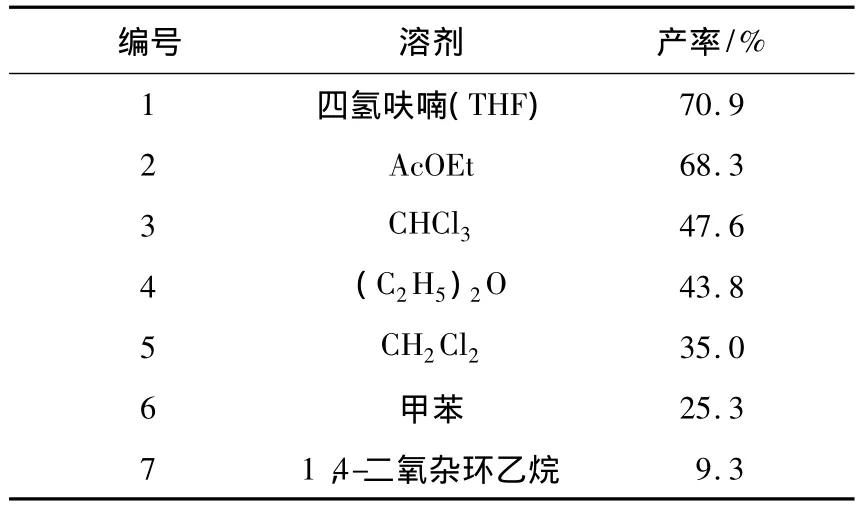
表2 溶剂对西司他丁酯产率的影响
2.2.3 温度的影响
反应温度对西司他丁酯的产率有着比较大的影响.以 THF 为溶剂,原料 7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ),半胱氨酸甲酯盐酸盐(Ⅵ)和碱的摩尔比为1∶1.5∶4,K3PO4为碱,考察了反应温度对产物产率的影响,结果见表3.从表3可以看出:当温度升高时,产物的产率有所提高;但过高的温度下,产物的产率反而下降.从表3得出,在80℃时产物的产率最高,为 74.1%.
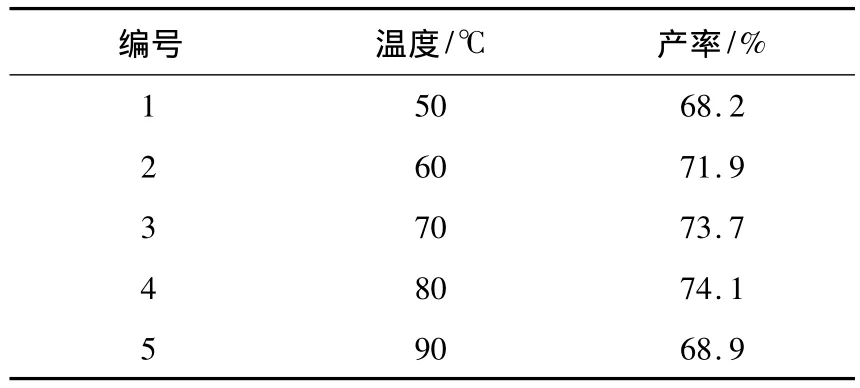
表3 温度对西司他丁酯产率的影响
2.2.4 碱用量的影响
碱的用量对西司他丁酯产率的影响较大.以THF 为溶剂,原料 7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ)和半胱氨酸甲酯盐酸盐(Ⅵ)的摩尔比为1 ∶1.5,K3PO4为碱,80℃反应时,考察了碱的用量对西司他丁酯产率的影响.从表4可以看出:碱的用量多少影响着产物产率的高低.碱用量增加,反应体系的碱性增加,使得产物的产率下降;碱用量减少,使得反应体系的碱性变弱,产物的产率也降低;原料Ⅴ与碱的摩尔比为1∶4时,产物的产率较高,为74.1%.

表4 碱的用量对西司他丁酯产率的影响
2.2.5 原料Ⅵ和Ⅴ摩尔比的影响
以THF为溶剂,K3PO4为碱,原料Ⅴ与碱的摩尔比为1∶4时,考察了原料半胱氨酸甲酯盐酸盐(Ⅵ)和 7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ)的摩尔比对西司他丁酯产率的影响.从表5可以看出:在保持碱的用量不变的情况下,当原料半胱氨酸甲酯盐酸盐(Ⅵ)和7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ)的摩尔比增加时,产物的产率明显降低;当原料Ⅵ和Ⅴ的摩尔比减少时,产物的产率亦明显降低;当原料Ⅵ和Ⅴ的摩尔比增加时,相应地增加碱的用量,产物的产率才能增加.因此,原料Ⅵ,Ⅴ和碱的摩尔比为1.5∶1∶4时,产物的产率较高,达到74.1%.

表5 原料Ⅵ与Ⅴ的摩尔比对西司他丁酯产率的影响
3 结论
本实验以(R)-1,1'-联萘酚为拆分试剂,经过酯化、氨解,能够有效地拆分2,2-二甲基环丙烷甲酸,高产率地得到光学纯S构型的2,2-二甲基环丙烷甲酰胺,并几乎定量地回收得到联萘酚;以半胱氨酸甲酯盐酸盐(Ⅵ)与7-氯-2-((1S)-2,2-二甲基环丙烷甲酰胺)-2-庚烯酸乙酯(Ⅴ)反应,得到了合成西司他丁酯的较佳条件,即以四氢呋喃为溶剂,K3PO4作为碱,温度为80℃,原料Ⅵ,Ⅴ和碱的摩尔比为1.5∶1∶4时,西司他丁酯的产率较高,达到74.1%.该合成方法不但可以高产率地得到手性的环丙烷甲酰胺,拆分剂定量地回收,并且在硫醚化的合成步骤中以K3PO4作为碱,避免了使用强碱和无水反应体系,反应条件温和,操作简便.
[1]Frederick M K,Helmut K,Jon G S,et al.Thienamycin:development of imipenem-cilastatin[J].J Anitimicrob Agents Chemother,1983,12(Suppl D):1-35.
[2]Donald W G,Wallace T A,Louis B,et al.Inhibition of the mammalian beta-lactamase renal dipeptidase(dehydropeptidase-I)by(Z)-2-(acylamino)-3-substituted-propenoic acids[J].J Med Chem,1987,30(6):1074-1090.
[3]徐晓莉,王海山,吴剑波,等.西司他丁的合成[J].中国医药工业杂志,1994,25(2):51-54.
[4]石晓华,王海峰,陈新志,等.西司他丁合成新工艺[J].河北化工,2007,30(12):44-46.
[5]Meul T.Verfahren zur racematspaltung von 2,2-dimethylcyclopropancarbonsäure:EP,0474200A2[P/OL].1992-03-11.http://v3.espacenet.com/searchResults?NUM=EP0474200&DB=EPODOC&submitted=true&locale=en_EP&ST=number&compact=false.
[6]石晓华,周舞阗,陈新志.2,2-二甲基环丙烷甲酸的合成与拆分[J].高等学校工程学报,2005,3(19):384-387.
[7]金洁,杨细文,武燕彬,等.西司他丁中间体(+)-(S)-2,2-二甲基环丙羧酸的合成[J].中国新药杂志,2004,13(5):419-420.
[8]Meul T.Process for the resolution of 2,2-dimethylcyclopropane carboxylic acid racemate:EP,0461541[P/OL].1994-12-14.http://v3.espacenet.com/searchResults?NUM=EP0461541&DB=EPODOC&submitted=true&locale=en_EP&ST=number&compact=false.
[9]Meul T.Verfahren zur herstellung optisch aktiver carbonsäuren durch racematspaltung:CH,682485[P/OL].1993-09-30.http://v3.espacenet.com/searchResults?NUM=CH682485&DB=EPODOC&submitted=true&locale=en_EP&ST=number&compact=false.
[10]Fujita K,Minomura M,Koizumi J.Process for preparing optically active carboxylic acid derivative:JP,WO02022543A1[P/OL].2002-03-21.http://v3.espacenet.com/searchResults?NUM= WO2002022543&DB=EPODOC&submitted=true&locale=en_EP&ST=number&compact=false.
[11]Lee S S,Hadinoto S,Ying J Y.Improved enantioselectivity of immobilized chiral bisoxazolines by partial precapping of the siliceous mesocellular foam support with trimethylsilyl groups[J].Adv Synth Catal,2006,348,1248-1254.
[12]Lee S S,Ying J Y.Siliceous mesocellular foam-supported chiral bisoxazoline:Application to asymmetric cyclopropanation[J].J Mol Catal A,2006,256:219-224.
[13]Wang Meixiang,Feng Guoqiang,Zheng Qiyu.Nitrile and amide biotransformations for efficient synthesis of enantiopure gem-dihalocyclopropane derivatives[J].Adv Synth Catal,2003,345:695-698.
[14]Wang Meixiang,Feng Guoqiang,Zheng Qiyu.Synthesis of high enantiomeric purity gem-dihalocyclopropane derivatives from biotransformations of nitriles and amides[J].Tetrahedron:Asymmetry,2004,15:347-354.
[15]Kumar Y,Tyagi O D,Strvastava T K,et al.Process for the preparation of cilastatin:JP,WO03018544[P/OL].2003-03-06.http://v3.espacenet.com/searchResults?NUM=WO03018544&D B=EPODOC&submitted=true&locale=en_EP&ST=number&compact=false.
猜你喜欢
上海计量测试(2020年1期)2020-03-18
天然气化工—C1化学与化工(2019年5期)2019-12-06
浙江化工(2019年4期)2019-05-13
农药科学与管理(2019年10期)2019-04-20
浙江大学学报(工学版)(2016年2期)2016-06-05
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
药学研究(2015年11期)2015-12-19
中国塑料(2015年4期)2015-10-14
中国药理学通报(2014年2期)2014-05-09
