
微波加热分解法制备CuO-Ce0.6Zr0.4O2及其催化CO氧化性能
2011-11-30 10:41杨志强毛东森吴仁春
物理化学学报 2011年5期
杨志强 毛东森 吴仁春 俞 俊 王 倩
(上海应用技术学院,化学与环境工程学院,上海200235)
微波加热分解法制备CuO-Ce0.6Zr0.4O2及其催化CO氧化性能
杨志强 毛东森*吴仁春 俞 俊 王 倩
(上海应用技术学院,化学与环境工程学院,上海200235)
采用微波加热分解法制备了一系列CuO含量(0-25%,w)不同的CuO-Ce0.6Zr0.4O2催化剂,并用X射线衍射(XRD)、透射电镜(TEM)、N2吸附-脱附(BET)和H2程序升温还原(H2-TPR)技术对催化剂进行了表征,利用色谱流动法考察了其催化CO低温氧化性能.结果表明:当CuO含量为15%时,所制备催化剂的催化活性最佳.在催化剂中存在三种不同类型的铜物种,即高分散、小颗粒和大颗粒的CuO.催化剂表面高分散和小颗粒的CuO有利于催化活性的提高,而大颗粒的CuO对催化活性具有抑制作用.
微波加热;氢氧化四氨合铜;CuO-Ce0.6Zr0.4O2催化剂;CO氧化
1 引言
CO的低温催化氧化在许多方面都有重要的应用价值.1,2目前,用于CO低温氧化的催化剂主要包括贵金属催化剂和贱金属催化剂两大类.由于贵金属资源稀少且价格昂贵,因此开发具有优良催化性能的贱金属催化剂成为近期研究的一大热点.其中, CuO/CeO2复合氧化物因具有优良的催化活性而受到广泛的关注.3,4近年来,CuO/CeO2-ZrO2催化剂因比CuO/CeO2具有更高的热稳定性5-7和更长的使用寿命7而得到了大量的研究.5-22目前,CuO/CeO2-ZrO2催化剂的制备方法可分为两类:一类是采用共沉淀、溶胶-凝胶、水热晶化和微乳等方法先制得CeO2-ZrO2复合氧化物,然后再采用浸渍法将CuO负载在CeO2-ZrO2复合氧化物的表面得到CuO/ CeO2-ZrO2催化剂,即所谓的两步法.5,7-18另一类是采用柠檬酸络合法、柠檬酸溶胶-凝胶法或表面活性剂模板法直接得到CuO-CeO2-ZrO2催化剂,6,17-22即所谓的一步法.
研究表明,CuO含量对CuO/CeO2-ZrO2催化剂的CO氧化活性具有很大的影响,而且不同作者所得到的结论并不相同.一种观点认为,CuO含量存在最佳值,即多了反而不好,具有副作用;6,8-10,14,15,22另一种观点认为,CuO含量多了无影响.19-21此外,洪庆红等17,18分别采用柠檬酸溶胶-凝胶一步法和柠檬酸溶胶-凝胶+浸渍两步法制备了CuO/Ce0.7Zr0.3O2催化剂,并考察了它们对CO氧化反应的催化活性.结果表明,当CuO含量为2.5%(w,下同)时,一步法所制备催化剂的活性明显大于两步法;而当CuO含量为15%时,两步法所制备催化剂的活性高于一步法.该结果表明,CuO含量对CuO/CeO2-ZrO2催化活性的影响与催化剂的制备方法密切相关.
最近,我们采用微波加热分解法(一步法)和微波加热处理共沉淀+浸渍法(两步法)制备了CuO/ CeO2-ZrO2催化剂.结果表明,一步法比两步法更有利于使催化剂表面CuO高度分散,CuO与CeO2-ZrO2间的相互作用更强,CuO更容易还原,从而具有更高的催化CO氧化活性.23在此基础上,本文采用微波加热分解法制备了一系列不同CuO含量(0-25%)的CuO-Ce0.6Zr0.4O2催化剂,并用X射线衍射(XRD)、透射电镜(TEM)、N2吸附-脱附(BET)和H2程序升温还原(H2-TPR)技术对催化剂进行了表征,考察了CuO含量对CuO-Ce0.6Zr0.4O2催化CO低温氧化性能的影响并与表征结果相关联.
2 实验部分
2.1 催化剂的制备
采用微波加热分解一步法制备CuO-Ce0.6Zr0.4O2催化剂.将准确称量的(NH4)2Ce(NO3)6、Zr(NO3)4· 5H2O和Cu(NO3)2·3H2O溶于适量的去离子水中,搅拌均匀,使溶液中金属离子的总浓度为0.1 mol·L-1,其中n(Ce):n(Zr)=3:2.采用反滴法,在快速搅拌下将上述混合液缓慢滴入到盛有稀氨水的三口烧瓶中.滴加结束,保证pH=10左右,并继续搅拌1 h.之后将反应混合物放入微波炉中,在快速搅拌下,微波加热回流,铜氨络合物分解.当pH降低至7时,继续微波加热10 min.稍冷后将沉淀母液抽滤,分别用去离子水和无水乙醇洗涤滤饼.然后把滤饼放入微波炉中加热干燥3 min,再在马弗炉中于600°C下焙烧2 h,制得CuO-Ce0.6Zr0.4O2催化剂,其中CuO含量分别为0,5%,10%,15%,20%和25%.上述所用试剂均为分析纯,由国药集团上海化学试剂有限公司生产.
2.2 催化剂的表征
XRD分析在荷兰X′Pert Pro型X射线衍射仪上进行.采用Cu Kα辐射(λ=0.15418 nm),管电流和管电压分别为40 mA和40 kV,扫描范围为20.0°-90.0°,精度为0.02°,扫描速率为8(°)·min-1.根据X射线衍射线宽法并利用Scherrer公式计算粒子的平均粒径.
采用JEM-2100(JEOL)型透射电子显微镜观察样品的晶粒大小及形貌,样品先在乙醇中超声分散,然后沉积在涂有碳膜的铜网上进行测试.
催化剂的比表面积采用美国Micromeritics公司生产的ASAP-2020M+C型自动物理化学吸附仪进行测定,样品先于200°C下抽真空脱气处理4 h,以高纯N2为吸附质,在液氮温度(-196°C)下进行测定.
催化剂的H2-TPR表征在常压微型石英管反应器中进行,样品的用量为50 mg,以10%H2+90%N2的混合气为还原气,气体流速为50 mL·min-1,升温速率为10°C·min-1.升温过程中H2的消耗量用浙江温岭福立公司生产的GC 9750型气相色谱仪的热导池检测器进行检测,检测器的温度为60°C.
2.3 催化剂的活性评价
CO氧化反应在常压微型固定床流动反应装置上进行.将0.2 g催化剂颗粒(40-60目)用0.6 g的石英砂均匀稀释后,装入下端塞有石棉布的内径为6 mm的不锈钢反应器中.在温度为200°C时用N2气(流速为45 mL·min-1)吹扫30 min,降至一定温度,然后切换为反应混合气(体积分数为1%CO、10%O2,其余为N2),流速为45 mL·min-1.反应前后的气体组成用浙江温岭福立公司生产的GC9790A型气相色谱仪进行在线分析,氢气为载气,热导池检测器,进样口和检测器的温度均为110°C,色谱柱为2 m×3 mm的TDX-01碳分子筛不锈钢填充柱,柱温采用一阶程序升温,初始温度40°C,保持5 min后以30°C· min-1的速率升温至130°C,然后保持5 min.
3 结果与讨论
3.1 XRD分析
图1是不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的XRD图.从中可以看出,所有样品均在2θ=29.2°, 33.8°,48.6°和57.8°处出现了CeO2的特征衍射峰,并且没有检测到ZrO2的特征衍射峰,表明所制备的催化剂中的CeO2和ZrO2形成了呈莹石结构立方相体系的复合氧化物.24

图1 不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的XRD图Fig.1 XRD patterns of CuO-Ce0.6Zr0.4O2catalysts with different CuO contentswCuO/%:(a)0,(b)5,(c)10,(d)15,(e)20,(f)25
根据CeO2(111)晶面的衍射峰利用Scherrer公式计算CeO2的平均粒径,结果如表1所示.从表1看出,CuO含量从0增加到15%时,CeO2晶粒的粒径有逐渐减小的趋势;当CuO含量从15%继续增加到25%时,CeO2晶粒的粒径基本不变.这说明CuO对铈锆复合氧化物的晶化和晶粒生长有抑制作用,随着CuO负载量从5%增加到15%,这种抑制作用变得比较明显,再继续增加CuO含量,对晶化的抑制作用开始变得微弱.我们采用固相反应法制备CuO-CeO2催化剂时也发现了这一现象.25其原因在于通过微波加热,铜氨络合物分解生成的CuO均匀沉淀于CeO2-ZrO2复合氧化物颗粒的表面,在高温焙烧过程中,CuO颗粒阻止了铈锆复合氧化物颗粒之间的聚集,进而抑制其晶粒的生长.23
另外,由图1可见,当CuO含量小于15%时,没有出现CuO的特征衍射峰.CuO/CeO2-ZrO2催化剂中的CuO除以晶相存在外,还可能以三种不同的方式存在,即高度分散于铈锆复合氧化物的表面,以微小的CuO颗粒存在而无法被XRD所检测到,进入CeO2-ZrO2的晶格中形成了Cu-Ce-Zr-O固溶体.关于Cu是否进入CeO2-ZrO2的晶格形成Cu-Ce-Zr-O固溶体,目前尚存争议.Escribano等26采用共沉淀+浸渍两步法制备的CuO含量为6.5%的CuO/ Ce0.5Zr0.5O2催化剂,与载体Ce0.5Zr0.5O2相比,其XRD衍射峰并未发生位移,表明Cu未进入CeO2-ZrO2的晶格.蔡超和薛屏27采用柠檬酸溶胶-凝胶一步法制备了Cu:Ce:Zr原子比为12:12:1的CuO-CeO2-ZrO2催化剂,添加Cu使CeO2-ZrO2衍射峰略向高角度偏移,表明部分铜离子进入CeO2-ZrO2晶格中生成了Ce-Zr-Cu-O三元固溶体氧化物,且由于Cu2+的半径(0.072 nm)小于Zr4+的半径(0.084 nm),当Cu2+取代部分Zr4+进入晶格时使晶格收缩.与此相反,郭锡坤等28采用同样方法制备的CuO-Ce0.2Zr0.8O2催化剂,与Ce-Zr-O固溶体相比衍射峰向小角度方向偏移,认为Cu+进入了Ce-Zr-O固溶体的晶格中形成了Cu-Ce-Zr-O固溶体,且由于Cu+的半径(0.096 nm)大于Ce3+的半径(0.092 nm)和Zr4+的半径(0.084 nm),所以使晶格增大,并随Cu含量的增加上述偏移越发明显.由图1中的局部放大图可见,本文所制备的CuO-CeO2-ZrO2催化剂与载体CeO2-ZrO2相比并未发现明显的位移,表明Cu未进入CeO2-ZrO2的晶格或进入的量极少可以忽略.当CuO含量达到15%后,在2θ为35.6°和38.8°处出现了单斜晶相CuO的特征衍射峰,而且随CuO含量的增加,其强度逐渐增强,尤其是当CuO含量从20%增加至25%时峰强度显著增大,这表明以晶相存在的CuO的颗粒逐渐增大.这与表1中采用Scherrer公式计算得到的CuO平均粒径的结果相一致.

表1 CuO-Ce0.6Zr0.4O2催化剂的比表面积(SBET)和CeO2及CuO的晶粒大小(D)Table 1 Specific surface areas(SBET)and crystal sizes(D) of CeO2and CuO of CuO-Ce0.6Zr0.4O2catalysts
3.2 TEM分析
图2为不同CuO含量CuO-CeO2-ZrO2催化剂的TEM照片.可以看出,图2(a)中样品呈接近球状颗粒,为铈锆复合氧化物的粒子,其粒径在6 nm左右,且未观察到CuO颗粒,这与上述XRD的表征结果相一致.图2(b)中大量的细小颗粒是铈锆复合氧化物的粒子,图中黑色的大颗粒物质为晶相的CuO,其粒径为40-50 nm,与用XRD计算的尺寸(表1)基本一致.此外,这些大颗粒的CuO均被小颗粒载体所包裹,这与我们之前采用两步法制备的CuO/Ce0.6Zr0.4O2催化剂中大颗粒CuO的存在方式不同,其CuO的颗粒均单独存在,未发现被载体所包裹的情况.23
3.3 BET比表面积测定

图2 不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的TEM照片Fig.2 TEM images of CuO-Ce0.6Zr0.4O2catalysts with different CuO contents wCuO/%:(a)5,(b)25
表1列出了不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的BET比表面积的测定结果.由表中数据可见,当CuO含量从0增加到10%时,样品的比表面积逐渐增大,而当负载量大于10%时,样品的比表面积逐渐降低.CuO对CuO-CeO2-ZrO2催化剂比表面积的影响存在两种不同的作用:一方面,CuO有抑制载体CeO2-ZrO2晶粒生长的作用,前述的XRD结果证明了这一点.随着CuO含量的增加,这种抑制作用增强,所以,催化剂的比表面积增大.另一方面,所添加的CuO覆盖在催化剂的表面或堵塞部分孔道,同时随着CuO含量的增加,CuO颗粒容易结晶长大,从而导致催化剂比表面积的降低.由上述结果可知,当CuO含量小于10%时,前一种作用起主要作用;而当CuO含量大于10%时,后一种作用起主要作用.
3.4 TPR分析
图3是CuO-Ce0.6Zr0.4O2催化剂的H2-TPR表征结果.由图3可见,CuO含量较低(≤10%)的CuOCe0.6Zr0.4O2催化剂具有两个还原峰(α和β),即存在两种不同类型的铜物种.随催化剂中CuO含量的增加,β峰的温度明显升高,从约200°C逐渐升高至215°C左右,而且峰强度逐渐增强、峰形变宽.对β峰进行曲线拟合发现其实存在2个峰,分别标记为β1和β2,其峰值温度分别为190和220°C.随催化剂中CuO含量的增加,α峰的温度基本不变,均在160°C左右.但强度先随CuO含量的增加而增强,至15%时达到最大,然后又随CuO含量的进一步增加而逐渐减弱.此外,随催化剂中CuO含量的增加,β1峰的强度逐渐减弱而β2峰的强度逐渐增强.
关于CuO/CexZr1-xO2催化剂中CuO还原性的研究已有不少报道.Wang等7在研究采用尿素水热晶化+浸渍两步法制备的CuO含量为5%的不同铈锆比的CuO/CexZr1-xO2催化剂时发现,在179、201和 245°C处存在3个还原峰,分别对应与载体强相互作用的高分散的CuO、与载体弱相互作用的较大颗粒的CuO和晶相CuO的还原.Ma等29也报道了采用柠檬酸溶胶-凝胶+浸渍两步法制备的CuO/ Ce0.8Zr0.2O2催化剂的H2-TPR谱中分别归属为高分散CuO、中等颗粒CuO和大颗粒CuO还原的3个耗氢峰.最近,Ayastuy等16采用浸渍法制备的CuO/CeO2-ZrO2催化剂分别在<100°C、123-136°C和176-195°C存在α、β和γ三个还原峰,分别为与载体相互作用的高分散CuO、与载体弱相互作用的CuO或孤立的Cu2+和大颗粒的体相CuO的还原峰.此外,我们之前采用微波加热处理共沉淀+浸渍两步法制备的CuO/Ce0.6Zr0.4O2催化剂分别在161-172°C、199-218°C和223-262°C出现3个归属为与载体强相互作用的高分散CuO的还原峰,与载体弱相互作用的小颗粒CuO的还原峰以及晶相CuO的还原峰.23根据上述文献及前述的XRD表征结果,我们认为本文CuO-Ce0.6Zr0.4O2催化剂中的α峰应归属于催化剂表面高度分散CuO的还原,β1峰应归属于催化剂表面较小颗粒(XRD无法检测)CuO的还原,β2峰应归属于催化剂表面较大颗粒(XRD能够检测)CuO的还原.
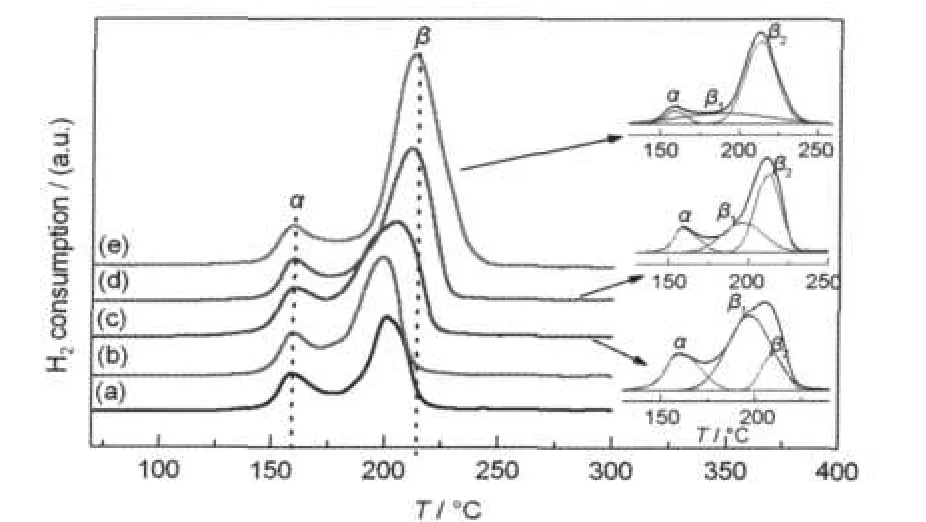
图3 不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的H2-TPR曲线Fig.3 H2-TPR curves of CuO-Ce0.6Zr0.4O2catalysts with different CuO contentswCuO/%:(a)5,(b)10,(c)15,(d)20,(e)25
3.5 催化剂的活性
图4为不同CuO含量的CuO-Ce0.6Zr0.4O2催化剂对CO氧化反应的催化活性.由此可见,随着反应温度的升高,所有催化剂上的CO转化率均逐渐提高.以CO转化率为90%时反应温度(T90)的高低来表示催化活性的大小,不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的T90值分别为89°C(5%)、83°C(10%)、80°C (15%)、85°C(20%)和87°C(25%).从中可以看出,随着催化剂中CuO含量的增加,CO的转化率先逐渐增大,至15%时达到最大,之后继续增加催化剂中CuO的含量,CO的转化率反而又开始下降.上述结果说明催化剂中CuO含量的增加并不一直有利于催化剂活性的提高,当CuO含量超过一定的值后,反而对催化剂的活性具有抑制作用,该结论与其它一些研究者所得到的结论相一致.6,8,-11,14,15,18-21蒋晓原等8,9采用共沉淀+浸渍两步法制备CuO/Ce0.5Zr0.5O2催化剂时发现CO催化活性先随CuO负载量的增加而增大,至5%-10%时达到最大,之后又开始下降. Wang等10采用同样方法制备的CuO/Ce0.8Zr0.2O2催化剂的活性在CuO含量为10%时达到最大值.Cao等22在研究采用表面活性剂模板法制备的介孔CuOCe0.8Zr0.2O2催化剂时发现,CO的氧化活性先随CuO含量(10%,15%,20%和25%)的增加而逐渐增大,至25%时达到最大,之后(30%和40%)反而降低.

图4 不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的CO氧化活性Fig.4 CO oxidation activity of CuO-Ce0.6Zr0.4O2catalysts with different CuO contentswCuO/%:(a)5,(b)10,(c)15,(d)20,(e)25
研究表明,CuO/CeO2-ZrO2催化CO氧化反应的活性与其CuO的存在形式密切相关.蒋晓原、8,9Wang10和Zhu15等都认为催化剂表面高分散的CuO是反应的活性中心,而体相的CuO具有副作用.此外,Chen等14认为催化剂表面高分散的CuO及小颗粒的CuO是CO低温氧化反应的活性中心.在本文中,将不同CuO含量CuO-Ce0.6Zr0.4O2催化剂的CO氧化活性与其XRD、TEM和TPR表征结果相关联可知,当CuO含量小于15%时,随CuO含量的增加,以高分散和小颗粒存在的CuO量增多,致使催化剂的活性也相应提高.当CuO含量增加到15%时,高分散和小颗粒存在的铜物种含量最多,故CO的氧化活性最高.之后随着CuO含量的继续增大,CuO的颗粒逐渐增大,使高分散和小颗粒存在的CuO的量减少,而大颗粒的CuO量增大,从而使催化剂的活性反而降低.这与Zhu等15的结果相一致,他们发现当CuO含量低于分散阈值时,CuO/Ce0.67Zr0.33O2催化剂的活性随CuO含量的增加而增大,当CuO含量明显高于分散阈值时,催化剂的活性显著降低.
此外,为了解本方法所制备CuO-Ce0.6Zr0.4O2催化剂的稳定性,我们对催化活性最高的CuO含量为15%的CuO-Ce0.6Zr0.4O2催化剂做了寿命实验.在反应温度控制在80°C的情况下持续反应48 h,CO的转化率基本维持不变.上述结果表明采用微波加热分解法所制备的CuO-Ce0.6Zr0.4O2催化剂具有非常高的活性和稳定性.
4 结论
CuO含量对微波加热分解法所制备的CuOCe0.6Zr0.4O2催化剂的催化性能产生显著的影响,其最佳值为15%.低CuO含量的催化剂存在高分散和小颗粒两种不同类型的铜物种,而高CuO含量的催化剂存在高分散、小颗粒和大颗粒(晶相)三种不同类型的铜物种.催化剂表面高分散和小颗粒的CuO有利于催化活性的提高,而大颗粒的晶相CuO对催化活性具有抑制作用.
(1) Liu,Y.;Meng,M.;Yao,J.S.;Zha,Y.Q.Acta Phys.-Chim.Sin. 2007,23,641.[刘 咏,孟 明,姚金松,査宇清.物理化学学报,2007,23,641.]
(2)Yu,J.;Wu,G.S.;Mao,D.S.;Lu,G.Z.Acta Phys.-Chim.Sin. 2008,24,1751.[俞 俊,吴贵升,毛东森,卢冠忠.物理化学学报,2008,24,1751.]
(3)Zheng,X.C.;Wang,X.Y.;Yu,L.H.;Wang,S.R.;Wu,S.H. Prog.Chem.2006,18,159.[郑修成,王向宇,于丽华,王淑荣,吴世华.化学进展,2006,18,159.]
(4) Liang,F.X.;Zhu,H.Q.;Qin,Z.F.;Wang,G.F.;Wang,J.G. Prog.Chem.2008,20,1453.[梁飞雪,朱华青,秦张峰,王国富,王建国.化学进展,2008,20,1453.]
(5)Wang,S.P.;Zheng,X.C.;Wang,X.Y.;Wang,S.R.;Zhang,S. M.;Yu,L.H.;Huang,W.P.;Wu,S.H.Catal.Lett.2005,105, 163.
(6)Wang,S.P.;Wang,X.Y.;Huang,J.;Zhang,S.M.;Wang,S.R.; Wu,S.H.Catal.Commun.2007,8,231.
(7)Wang,S.P.;Zhang,T.Y.;Su,Y.;Wang,S.R.;Zhang,S.M.; Zhu,B.L.;Wu,S.H.Catal.Lett.2008,121,70.
(8) Jiang,X.Y.;Zhou,R.X.;Chen,Y.;Lou,L.P.;Zheng,X.M. J.Zhejiang Univ.:Science Edition 2001,28,653. [蒋晓原,周仁贤,陈 煜,楼莉萍,郑小明.浙江大学学报:理学版, 2001,28,653.]
(9)Jiang,X.Y.;Chen,Y.;Zhou,R.X.;Zheng,X.M.J.Fuel Chem. Technol.2001,29(Suppl.),122.[蒋晓原,陈 煜,周仁贤,郑小明.燃料化学学报,2001,29(增刊),122.]
(10)Wang,S.P.;Wang,X.Y.;Zheng,X.C.;Wang,S.R.;Zhang,S. M.;Huang,W.P.;Wu,S.H.React.Kinet.Catal.Lett.2006,89, 37.
(11)Luo,M.F.;Zheng,X.M.Acta Chem.Scand.1998,52,1183.
(12)Yang,Z.Q.;Mao,D.S.;Zhu,H.L.;Lu,G.Z.Chin.J.Catal. 2009,30,997.[杨志强,毛东森,朱慧琳,卢冠忠.催化学报, 2009,30,997.]
(13) Martinez-Arias,A.;Fernandez-Garcia,M.;Galvez,O.; Coronado,J.M.;Anderson,J.A.;Conesa,J.C.;Soria,J.; Munuera,G.J.Catal.2000,195,207.
(14)Chen,H.L.;Zhu,H.Y.;Wu,Y.;Gao,F.;Dong,L.;Zhu,J.J. J.Mol.Catal.A 2006,255,254.
(15) Zhu,J.;Zhang,L.L.;Deng,Y.;Liu,B.;Dong,L.H.;Gao,F.; Sun,K.Q.;Dong,L.;Chen,Y.Appl.Catal.B 2010,96,449.
(16)Ayastuy,J.L.;Gurbani,A.;Gonzalez-Marcos,M.P.;Gutierrez-Ortiz,M.A.Appl.Catal.A 2010,387,119.
(17) Hong,Q.H.;Song,Y.P.;Jia,A.P.;Pu,Z.Y.;Luo,M.F.J.Mol. Catal.(China)2008,22,429.[洪庆红,宋宇鹏,贾爱平,普志英,罗孟飞.分子催化,2008,22,429.]
(18) Hong,Q.H.;Lin,H.;Song,Y.P.;Wu,C.S.J.Jinhua Coll. Prof.Technol.2009,9,49.[洪庆红,林 鸿,宋宇鹏,吴昌胜.金华职业技术学院学报,2009,9,49.]
(19) Wang,E.G.;Chen,S.Y.J.Chin.Rare Earth Soc.2001,19, 17. [王恩过,陈诵英.中国稀土学报,2001,19,17.]
(20)Wang,E.G.;Chen,S.Y.J.Rare Earths 2002,20,533.
(21)Wang,J.P.;Wang,E.G.;Chen,S.Y.Coal Conv.2000,23,88. [王建平,王恩过,陈诵英.煤炭转化,2000,23,88.]
(22)Cao,J.L;Wang,Y.;Zhang,T.Y.;Wu,S.H.;Yuan,Z.Y.Appl. Catal.B 2008,78,120.
(23)Yang,Z.Q.;Mao,D.S.;Guo,Q.S.;Gu,L.Acta Phys.-Chim. Sin.2010,26,3278.[杨志强,毛东森,郭强胜,顾 蕾.物理化学学报,2010,26,3278.]
(24)Yang,Z.Q.;Mao,D.S.;Zhu,H.L.;Lu,G.Z.Chin.J.Inorg. Chem.2009,25,812.[杨志强,毛东森,朱慧琳,卢冠忠.无机化学学报,2009,25,812.]
(25)Mao,D.S.;Tao,L.H.;Wang,Q.;Guo,Y.L.;Lu,G.Z.Chin.J. Inorg.Chem.2010,26,447.[毛东森,陶丽华,王 倩,郭杨龙,卢冠忠.无机化学学报,2010,26,447.]
(26) Escribano,V.S.;Martínez,C.H.;López,E.F.;Amores,J.M. G.;Busca,G.Catal.Commun.2009,10,861.
(27) Cai,C.;Xue,P.J.Ningxia Univ.:Natural Science Edition 2005, 26,345.[蔡 超,薛 屏.宁夏大学学报:自然科学版, 2005,26,345.]
(28)Guo,X.K.;Jia,H.J.;Fan,Q.Y.;Li,W.Y.;Liu,W.M.Chin.J. Catal.2009,30,124.[郭锡坤,贾海建,范倩玉,李文衍,刘伟明.催化学报,2009,30,124.]
(29) Ma,L.;Luo,M.F.;Chen,S.Y.Appl.Catal.A 2003,242,151.
December 22,2010;Revised:February 14,2011;Published on Web:March 22,2011.
Preparation of CuO-Ce0.6Zr0.4O2by Microwave Heating Decomposition and Its Catalytic Property for CO Oxidation
YANG Zhi-Qiang MAO Dong-Sen*WU Ren-Chun YU Jun WANG Qian
(School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai 200235,P.R.China)
A series of CuO-Ce0.6Zr0.4O2catalysts with different contents(0-25%,w)of CuO were prepared by microwave heating decomposition and characterized by X-ray diffraction(XRD),transmission electron microscopy(TEM),N2adsorption-desorption(BET),and temperature-programmed reduction with hydrogen (H2-TPR).The activities of the catalysts for CO oxidation were evaluated using a microreactor-gas chromatograph system.The results show that the CuO-Ce0.6Zr0.4O2catalyst exhibits the best catalytic activity for CO oxidation at a CuO content of 15%.Three copper species are present in the catalysts,i.e., highly dispersed,small and large CuO particles.The highly dispersed and small CuO particles are responsible for the promotion of catalytic activity while the large CuO particles inhibit catalytic activity.
Microwave heating;Cu(NH3)4(OH)2;CuO-Ce0.6Zr0.4O2catalyst;CO oxidation
O643
*Corresponding author.Email:dsmao@sit.edu.cn,dsmao1106@yahoo.com.cn;Tel/Fax:+86-21-64941386.
The project was supported by the LeadingAcademic Discipline Project of Shanghai Education Committee,China(J51503).上海市教委重点学科建设项目(J51503)资助
猜你喜欢
化工进展(2022年5期)2022-05-26
陕西科技大学学报(2021年5期)2021-10-27
陶瓷学报(2020年6期)2021-01-26
云南化工(2020年11期)2021-01-14
中国粉体技术(2019年3期)2019-05-14
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年7期)2014-02-28
无机化学学报(2014年4期)2014-02-28
