
固体酸催化合成ε-己内酯的研究Ⅱ.合成工艺条件的研究
2011-11-09 06:03:08欧华强张光旭胡昌林徐文涛胡张雁
石油化工 2011年8期
欧华强,张光旭,胡昌林,徐文涛,胡张雁
(武汉理工大学 化学工程学院,湖北 武汉 430070)
固体酸催化合成ε-己内酯的研究Ⅱ.合成工艺条件的研究
欧华强,张光旭,胡昌林,徐文涛,胡张雁
(武汉理工大学 化学工程学院,湖北 武汉 430070)
采用混烧法制备了B2O3/γ-Al2O3催化剂,并将其用于以H2O2为氧化剂催化氧化环己酮两步法合成ε-己内酯(ε-CL)。利用单因素实验考察了第一步反应中反应压力、原料配比、催化剂用量和反应时间对过氧丙酸收率和选择性的影响;同时考察了第二步反应中反应温度和时间对ε-CL的收率和选择性的影响。实验结果表明,在第一反应中,当反应压力48.13 kPa、n(丙酸)∶n(H2O2)=4∶1、B2O3/γ-Al2O3催化剂用量0.8 g、反应时间3 h时,过氧丙酸的收率和选择性分别可达到17.08%和27.51%;在第二步反应中,当环己酮用量5.40 g、常压、反应温度50℃、反应时间3 h时,ε-CL的收率和选择性分别可达到87.72%和95.07%。
固体酸催化剂;氧化硼/氧化铝催化剂;过氧化氢;环己酮;ε-己内酯
ε-己内酯(ε-CL)是一种重要的有机合成中间体,主要用于合成聚己内酯,也可与其他酯类共聚或共混改性。共聚或共混改性得到的聚合物具有良好的热塑性、成型加工性、生物相容性、无毒性、药物透过性和生物降解性,在材料、环保和医用等方面具有广泛的应用前景[1-5],使ε-CL的合成研究愈加重要。国内外有关ε-CL合成方法的报道很多[6-15],包括过氧酸氧化法、H2O2氧化法、O2/空气氧化法及生物氧化法等,其中以有机酸为介质、H2O2为氧化剂催化氧化环己酮合成ε-CL的方法最为人们所关注。但该方法存在催化剂活性低、ε-CL的收率和选择性差、后续过程产品不易分离及催化剂难以回收等问题。
本工作采用混烧法制备了B2O3/γ-Al2O3固体催化剂,并将其用于以H2O2为氧化剂催化氧化环己酮两步法合成了ε-CL。考察了两步反应中反应压力、原料配比、催化剂用量、反应时间和温度等对合成反应的影响,确定了最佳的合成工艺条件。
1 实验部分
1.1 反应原理
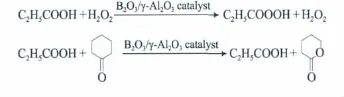
ε-CL的合成包括两步:第一步为过氧丙酸的制备,丙酸与H2O2在B2O3/γ-Al2O3催化剂作用下生成过氧丙酸和水,体系中的水与带水剂环己烷形成共沸物,水通过油水分离器排出体系;第二步为ε-CL的合成,第一步得到的过氧丙酸与环己酮在B2O3/γ-A l2O3催化剂作用下生成丙酸和ε-CL。两步反应的方程式为:
1.2 催化剂的制备与表征
将球形 γ-Al2O3研磨成粉末,将硼酸和 γ-Al2O3粉末按比例混合均匀,然后在300℃下焙烧3 h即得 B2O3/γ-Al2O3催化剂。对催化剂进行XRD、N2吸附-脱附、TEM和SEM等表征,表征结果见文献[16]。
1.3 ε-CL的合成
ε-CL的合成装置见图1。

图1 ε-CL的合成装置Fig.1 Experimental apparatus for ε-caprolactone(ε-CL)synthesis.
将0.8 g催化剂、35 g环己烷、1滴二甲基吡啶和65.19 g丙酸加入三颈烧瓶中,水浴温度保持65℃,在压力48.13 kPa下将25.00 g H2O2(质量分数30%)由恒压加料漏斗缓慢加入,保持环己烷回流,连续脱水3 h,水由油水分离器底部排出;然后在50℃、常压下缓慢加入5.40 g环己酮,回流反应2 h。
1.4 分析测试
1.4.1 定性分析
用Agilent公司6890N/5973N型气相色谱-质谱联用仪对合成的ε-CL进行定性分析。GC分析条件:He为载气,FID检测,进样温度260℃,柱温采用程序升温(50℃,保留2 min,以10℃/min的速率升至120℃并保留2 min,再以30℃/m in的速率升至250℃并保留2 min),采用分流进样,分流比为50∶1。MS分析条件:离子源温度230℃,GC和MS接口温度150℃,扫描m/z=19~750。在上述条件下,将试样提取液用Scan方式进行GC-MS分析。
1.4.2 定量分析
利用滴定分析法[17]定量分析第一步反应液中的H2O2和过氧丙酸;利用Agilent公司6890N型气相色谱仪通过外标法对第二步反应液中的环己酮和ε-CL进行定量分析。GC分析条件:HP-5毛细管色谱柱(30 m×0.32 mm×0.25μm),N2为载气,FID检测,N2流量30 L/min,H2流量30m L/min,空气流量300 m L/min,进样器温度250℃,柱温采用程序升温(80℃,保留1 min,以10℃/min的速率升至120℃并保留1 min,再以30℃/min的速率升至180℃并保留2 min),采用分流进样,分流比为50∶1。
2 结果与讨论
2.1 ε-CL的MS分析结果
合成的ε-CL和ε-CL标准试样的MS谱图见图2。由图2可知,合成的ε-CL的MS谱图和标准试样的MS谱图相比,具有很高的匹配度,根据有机质谱解析基本原理推测ε-CL可能发生以下断裂并生成相应离子碎片:m/z=114处的分子离子C6H10O+2首先发生α-断裂开环,然后进行McLafferty重排,脱掉中性小分子,同时生成 C4H8CO+(m/z=84),C3H6CO+(m/z=70),C4H+7(m/z= 55),C3H+6(m/z=42)等分子离子。通过以上分析可知,合成的化合物结构与ε-CL的结构基本一致。
2.2 第一步合成工艺条件的考察
2.2.1 反应压力的影响
反应压力对过氧丙酸收率和选择性的影响见图3。由图3可见,在第一步反应中,随反应压力的升高,过氧丙酸收率先增大后减小。当反应压力较低时,虽然分水效果较好,但H2O2与过氧丙酸容易被抽出,使分水器中水相中所含的过氧丙酸含量增加,从而导致过氧丙酸的收率下降。当反应压力较高时,一方面反应的分水效果变差,不利于H2O2向过氧丙酸转化;另一方面,反应压力较高会造成体系的共沸温度过高,导致H2O2分解较多,因此过氧丙酸收率很低。随反应压力的升高,过氧丙酸的选择性逐渐降低,这是因为反应压力越高,体系共沸温度越高,过氧化物分解越多。综合考虑,选择第一步反应压力为48.13 kPa较适宜。

图2 合成的ε-CL(a)和ε-CL标准试样(b)的MS谱图Fig.2 MS spectra of ε-CL product(a)and standard sample(b).

图3 反应压力对过氧丙酸收率和选择性的影响Fig.3 Effects of pressure on yield and selectivity of per-propionic acid in the first step.
2.2.2 原料配比的影响
n(丙酸)∶n(H2O2)对过氧丙酸收率和选择性的影响见图4。由图4可见,随n(丙酸)∶n(H2O2)的增大,过氧丙酸收率和选择性都呈现先增大后减小的趋势。当丙酸用量较少时,H2O2反应不充分,过氧丙酸收率很低;当n(丙酸)∶n(H2O2)=4∶1时,过氧丙酸收率和选择性均达到最大;当丙酸大量过量(即n(丙酸)∶n(H2O2)=6∶1)时,过氧丙酸收率很低,这是由于丙酸溶剂大量过量使反应体系浓度稀释所致。因此,选择n(丙酸)∶n(H2O2)= 4∶1较适宜。

图4 n(丙酸)∶n(H2O2)对过氧丙酸收率和选择性的影响Fig.4 Effects of n(propionic acid)∶n(H2 O2)on the yield and selectivity of per-propionic acid in the first step.
2.2.3 催化剂用量的影响
催化剂用量对过氧丙酸收率和选择性的影响见图5。由图5可见,当催化剂用量较少时,随催化剂用量的增加,过氧丙酸的收率和选择性均增大;当催化剂用量大于0.8 g时,随催化剂用量的增加,过氧丙酸收率增幅不大,选择性反而有所降低。这是因为,在催化剂用量较少时,随催化剂用量增加,催化活性中心数量增多,过氧丙酸的合成速率加快。但当催化剂用量过多时,反应液黏度增大,不利于催化反应传质,且反应的副产物也增加。因此,选择催化剂用量为0.8 g较适宜。

图5 催化剂用量对过氧丙酸收率和选择性的影响Fig.5 Effects of B2 O3/γ-Al2 O3 catalyst dosage on the yield and selectivity of per-propionic acid in the first step.
2.2.4 反应时间的影响
反应时间对过氧丙酸收率和选择性的影响见图6。

图6 反应时间对过氧丙酸收率和选择性的影响Fig.6 Effects of reaction time on the yield and selectivity of per-propionic acid in the first step.
由图6可见,在第一步反应中,随反应时间的延长,过氧丙酸收率呈现先增大后减小的趋势;在反应进行到3 h时,过氧丙酸收率达到最大。这是因为随反应时间的延长,H2O2氧化丙酸迅速转化生成过氧丙酸,使过氧丙酸收率逐渐增大,但随反应时间的进一步延长,H2O2浓度降低,生成的过氧丙酸量越来越少,另一方面反应时间过长,过氧丙酸的分解较多,因此过氧丙酸的收率降低。随反应时间的延长,过氧丙酸的选择性降低,这是由过氧丙酸持续分解所致。考虑到在保证过氧丙酸收率较高时,缩短反应时间对生产有利,因此选择第一步反应时间为3 h较适宜,此时过氧丙酸的收率和选择性分别达到17.08%和27.51%
2.3 第二步合成工艺条件的考察
2.3.1 反应温度的影响
反应温度对ε-CL收率和选择性的影响见图7。由图7可见,在第二步反应中,随反应温度的升高,ε-CL收率先增大后减小,ε-CL选择性呈下降趋势。这是因为随反应温度的升高,催化剂活性增强,有利于过氧丙酸氧化环己酮合成ε-CL。但当反应温度过高时副反应速率大大加快,副产物相应增多,导致ε-CL收率和选择性降低。因此,选择第二步的反应温度为50℃较适宜。

图7 反应温度对ε-CL收率和选择性的影响Fig.7 Effects of reaction temperature on the yield and selectivity ofε-CL in the second step.
2.3.2 反应时间的影响
反应时间对ε-CL收率和选择性的影响见图8。由图8可见,在第二步反应中,随反应时间的延长,ε-CL收率逐渐增大,反应初期收率增幅较大,反应超过2 h后收率增幅逐渐趋缓。随反应时间的延长,ε-CL选择性逐渐降低,且降幅越来越大。这是由于反应时间较长时,环己酮充分转化成为ε-CL,ε-CL收率增大。同时,随反应时间的延长,产生大量副产物,因此ε-CL选择性急剧下降。综合考虑,选择第二步反应时间为3 h较适宜,此时ε-CL的收率和选择性分别达到 87.72%和 95.07%。

图8 反应时间对ε-CL收率和选择性的影响Fig.8 Effects of reaction time on the yield and selectivity of ε-CL in the second step.
3 结论
(1)采用混烧法制备了 B2O3/γ-Al2O3催化剂,用于催化反应两步合成ε-CL。该催化剂具有较高的催化活性和选择性。
(2)由单因素实验得到两步催化反应合成ε-CL的优化条件为:第一步反应过程中,当反应压力48.13 kPa、n(丙酸)∶n(H2O2)=4∶1、B2O3/γ-Al2O3催化剂用量0.8 g、反应时间3 h时,过氧丙酸的收率和选择性分别达到17.08%和27.51%,过氧丙酸的收率和选择性偏低是由H2O2及过氧丙酸分解所致;第二步反应过程中,在环己酮用量5.40 g、常压、反应温度50℃、反应时间3 h时,ε-CL的收率和选择性分别达到87.72%和95.07%。
[1] Guerra G D,Cerrai P,Tricoli M,et al.Release of 5-Fluorouracil by Biodegradable Poly(Ester-Ether-Ester)s.Part I:Release by Fused Thin Sheets[J].J Mater Sci:Mater Med,2001,12(4):313-317.
[2] 杨安乐,孙康,吴人杰.聚ε-己内酯的合成、改性和应用进展[J].高分子通报,2000,6(2):52-57.
[3] 王宁,于九皋,马骁飞.生物降解热塑性材料的研究进展[J].石油化工,2007,36(1):1-8.
[4] 朱蔚璞,陈伟,沈之荃.三(2,6-二叔丁基-4-甲基苯氧基)镧配合物催化合成星形聚己内酯[J].催化学报,2007,28 (6):547-550.
[5] 易国斌,王永亮,康正,等.聚乙烯基吡咯烷酮对聚乙烯基吡咯烷酮/聚己内酯半互穿网络水凝胶性能的影响[J].石油化工,2008,37(2):183-186.
[6] 栗洪道.ε-己内酯的合成研究[J].江苏石油化工学院学报,2002,14(2):11-13.
[7] 杜宗罡,朱光明,於秋霞.ε-己内酯的合成及应用[J].化工新型材料,2003,31(9):12-14,18.
[8] Arnold L,Elings JA,Macquarrie D J,et al.The Baeyer-Villiger Oxidation of Ketones Using HMS-Supported Peroxycarboxylic Acids[J].Synlett,2000,2000(7):1052-1054.
[9] Mandal D,Ahmad A,Kham M I.Biocatalytic Transformation of Cyclohexanone by Fusarium SP[J].J Mol Catal Chem,2002,181(122):237-241.
[10] 刘媛,陈长林,徐南平.负载锑的水滑石催化环己酮Baeyer-Villiger氧化制 ε-己内酯[J].催化学报,2004,25(10):801-804.
[11] 张萍,杨梅,吕效平.在超声波气升式内环流反应器中合成ε-己内酯[J].石油化工,2006,35(1):19-23.
[12] 程东恩,张光旭,陈应澈.环己酮氧化合成ε-己内酯的研究进展[J].武汉化工学院学报,2006,28(4):31-34.
[13] 黄靓,李静霞,戴维林,等.含锡催化剂在Baeyer-Villiger氧化反应中的应用进展[J].石油化工,2007,36(2):200-204.
[14] 李静霞,黄靓,戴维林,等.高活性MgO/SnO2复合金属氧化物催化剂的合成及其在双氧水选择氧化环己酮制ε-己内酯反应中的应用[J].化学学报,2008,66(1):5-9.
[15] 徐文涛,欧华强,张光旭,等.环己酮催化氧化合成ε-己内酯催化剂的研究——介孔分子筛超强酸催化剂的研制及表征[J].山东化工,2008,(10):1-4.
[16] 张光旭,欧华强,胡昌林,等.固体酸催化合成ε-己内酯的研究:Ⅰ.催化剂的表征及其催化性能[J].石油化工,2011,40 (5):486-491.
[17] 全国文献工作标准化技术委员会第七分委员会.GB/T 19108—2003过氧乙酸溶液 过氧乙酸含量的测定[S].北京:中国标准出版社,2003.
Synthesis of ε-Caprolactone over Solid Acid Catalysts Ⅱ.Study on the Synthesis Conditions
Ou Huaqiang,Zhang Guangxu,Hu Changlin,Xu Wentao,Hu Zhangyan
(College of Chemical Engineering and Technology,Wuhan University of Technology,Wuhan Hubei430070,China)
B2O3/γ-Al2O3catalyst was prepared by co-calcination method and used as the catalyst in a two-step synthesis of ε-caprolactone(ε-CL)from cyclohexanone and H2O2oxidant.Effects of reaction pressure,n(propionic acid)∶n(H2O2),catalyst dosage and reaction time on yield and selectivity of perpropionic acid(intermediate product)in the first step reaction,and effects of reaction temperature and reaction time on yield and selectivity of ε-CL in the second step reaction were investigated.The results indicated that,in the first step,under reaction pressure 48.13 kPa,n(propionic acid)∶n(H2O2)4∶1,catalyst dosage 0.8 g and reaction time 3 h,the yield and selectivity of perpropionic acid could be 17.08%and 27.51%respectively,and in the second step,under cyclohexanone 5.40 g,normal pressure,reaction temperature 50℃ and reaction time 3 h,the yield and selectivity ofε-CL could reach 87.72%and 95.07%respectively.
solid acid catalyst;boron oxide/alum ina catalyst;hydrogen peroxide;cyclohexanone; ε-caprolactone
1000-8144(2011)08-0835-05
TQ 032.4
A
2011-02-14;[修改稿日期]2011-04-25。
欧华强(1985—),男,湖北省宜昌市人,硕士生,电话18622209753,电邮 ouhuaqiang@163.com。联系人:张光旭,电话13871467069,电邮zhanggx2002@163.com。
武汉理工大学自主创新研究基金项目(2010-ZYHG-004)。
(编辑 李明辉)
精细化工
猜你喜欢
化工管理(2021年7期)2021-05-13 00:45:22
中国粮油学报(2019年4期)2019-07-12 09:06:38
——过氧碳酸钠
农村青少年科学探究(2018年4期)2018-07-04 09:47:16
生物学教学(2016年4期)2016-08-15 00:43:11
现代食品(2016年24期)2016-04-28 08:12:06
石油炼制与化工(2016年4期)2016-04-06 20:19:54
合成化学(2015年10期)2016-01-17 08:55:57
化工进展(2015年3期)2015-11-11 09:07:41
医学研究杂志(2015年5期)2015-06-10 06:43:26
应用化工(2014年3期)2014-08-16 13:23:50
