
钴钼基水煤气变换催化剂及其催化反应工艺
2011-11-09 00:44连奕新杨意泉方维平
石油化工 2011年4期
连奕新,杨意泉,方维平
(厦门大学 化学化工学院 醇醚酯化工清洁生产国家工程实验室,福建 厦门 361005)
特约述评
钴钼基水煤气变换催化剂及其催化反应工艺
连奕新,杨意泉,方维平
(厦门大学 化学化工学院 醇醚酯化工清洁生产国家工程实验室,福建 厦门 361005)
介绍了国内外CO变换催化剂的发展及其在不同变换工艺中的应用,阐述了Co-Mo基催化剂的催化机理与硫化方法。通过综合分析中温变换、中温变换串联低温变换、中温变换串联两段低温变换和全程低温变换4种代表性的工艺流程,提出了高效节能型的全程低温变换工艺将是变换技术的发展方向,开发适应全程低温变换工艺的高活性和高稳定性的Co-Mo基变换催化剂及其预硫化技术是今后的主要研究目标。
水煤气变换;钴钼基催化剂;全程低温变换;一氧化碳;氢气;二氧化碳
自20世纪50年代以来,我国已掌握了以焦炭、无烟煤、焦炉气、天然气、油田伴生气和液态烃多种原料生产氨和尿素的技术,形成了特有的以煤、石油、天然气为原料,大、中、小生产规模并存的格局。由于石油价格飞涨,基于装置的经济性考虑,“轻油”和“重油”型合成氨装置已不具备市场竞争力,因而以煤为原料制氨等煤化工及其相关技术再度成为研发热点。煤炭作为我国的主要能源,在化学工业中占有十分重要的地位。近年来,我国煤基合成氨和合成甲醇产业的相关生产技术获得了全面进步[1-5]。
水煤气变换反应广泛应用于合成氨、制氢、合成气制醇和制烃等催化过程以及城市煤气中CO含量的调控。近年来,水煤气变换反应在燃料电池和生物质制氢技术中的应用越来越受重视[6-9]。在水煤气变换反应中,水蒸气是反应物,为了降低CO的含量并保证高变换率,水蒸气往往过量。水蒸气量的多少是衡量变换工艺能耗的重要标志,因此减少水蒸气量对工业节能降耗意义重大[10-11]。随着加压煤气化和合成氨生产大型化工艺的出现和发展,相应的变换工艺及其催化剂也有了很大的改进。国内外研究者都在竞相研究高效节能型变换工艺,其中最有效的节能措施就是降低变换反应的汽气比(蒸汽与干煤气的摩尔比),使变换工序在低汽气比下操作,因此开发适合低汽气比的催化剂成为研究重点。特别是各种新型节能流程相继开发成功,研制适合新工艺的节能型催化剂成为必然[12-13]。
1 变换催化剂的现状与发展
水煤气变换反应的研究始于1888年,Fe-Cr基催化剂在1913年研制成功,并在德国BASF公司合成氨厂首次实现工业化应用[10,14]。至今,水煤气变换反应在工业上应用已有上百年的历史。传统的变换催化剂通常指的是Fe-Cr基高温(350~520℃)变换催化剂,主要以尖晶石结构的Fe3O4为活性相、Cr2O3为主要助剂组成,亦称为Fe3O4-Cr2O3尖晶石固溶体。以不同制备方法得到的氧化铁为原料生产的催化剂,其活性亦不同。为提高催化剂性能,部分型号的催化剂中还添加了K2O,CaO,MgO或Al2O3等助剂。Fe-Cr基催化剂具有活性高、热稳定性好、寿命长和机械强度高等优点,但是此类催化剂在使用中需要大量的过剩水蒸气,以防止催化剂活性组分Fe3O4被过度还原成金属铁或碳化铁以及在低汽气比下发生费托合成副反应[15-16]。
从20世纪80年代以来,由于节能流程逐渐成为合成氨工业发展的主流,如凯洛格低能耗流程、ICI-AMV流程及布朗流程等,传统的高温变换催化剂已不能满足合成氨工业发展的要求,研制适合新工艺的节能型催化剂成为必然。为此世界各著名催化剂公司相继研制出适应低汽气比的改进型Fe-Cr基高温变换催化剂,如ICI71-4,SK-201,K6-11,C12-4 等[17-18]。此外作为 Fe-Cr基催化剂中主要稳定剂的Cr2O3,其毒性,特别是致癌性越来越引起了人们的高度关注。因此,研制出无Cr的Fe基或无Cr非Fe基的高温变换催化剂,降低催化剂在生产、使用过程中的能耗,成为该领域的主要研究方向[19-21]。
随后开发的活性温度较低(190~250℃)的Cu-Zn基变换催化剂,主要应用于以天然气为原料制合成气的CO低温变换工艺中,1963年首先在美国的合成氨工业中得到应用[22]。Cu-Zn基变换催化剂的主要活性组分是CuO,添加了ZnO,Al2O3或Cr2O3作为结构助剂,在催化剂制备过程中可形成热稳定性很高的Zn-Al或Cr-Al尖晶石,从而促进Cu微晶的分散。在过去的几十年,有大量关于Cu-Zn基低温变换催化剂制备方法、催化性能和机理的报道,同时Cu-Zn基低温变换催化剂组分也从最初的 Cu-Zn-Cr发展为 Cu-Zn-Al[23-24]。近年来,对于Cu-Zn低温变换催化剂的研究主要集中在探讨催化反应活性位的微观组成结构和反应机理以及通过改进其制备方法、添加一些助剂提高其耐热性[25-27]。但目前的研究结果表明,耐热性提高幅度有限,而且该类催化剂基本能够满足现有生产工艺的要求,所以有关该类催化剂的研究报道逐渐减少。
从工艺方面看,最初使用的Fe-Cr基催化剂由于存在活性温度较高、水蒸气消耗量较大等缺点,同时受热力学平衡的限制,固定床绝热反应器出口CO体积分数仅能降低到2% ~3%,只能采用铜氨洗工艺脱除剩余的微量CO。如果在甲醇串联甲烷化流程中采用甲烷化法脱除剩余的微量CO,根据工艺要求,变换气中CO体积分数需降至0.3%以下,显然单独使用Fe-Cr基变换催化剂很难达到要求[19,28]。虽然随后开发的 Cu-Zn基低温变换催化剂的活性温度低,但其耐硫和耐热性能却极差,反应温度高于250℃后活性急剧下降。所以从工艺角度出发,Cu-Zn基低温变换反应器进口的CO体积分数不能超过5%,以避免由于热效应造成催化剂床层温升过大,这一点限制了它的使用范围[29]。
十几年来,随着原料路线多元化制气技术的发展,要求变换催化剂除具有高的活性外,还需要有良好的抗硫性能。与传统的Fe-Cr基和Cu-Zn基催化剂相比,Co-Mo基催化剂的特点是只有当活性组分处在硫化状态下才具有活性,因而不存在硫中毒问题,不需要预脱除原料气中的硫化物,而且具有操作弹性大、活性高、使用温区宽(180~500℃)、不易中毒和耐硫等优点。因此许多国家进行了Co-Mo基催化剂的研究开发,并于1978年首次实现工业化生产与应用[30-32]。随着合成氨和合成甲醇等煤基化工的飞速发展,国内外对Co-Mo基变换催化剂的研究越来越活跃,着重于研制具有活性高、机械强度高、稳定性好、再生能力强、宽汽气比、宽温、耐硫、节能、低成本和制备工艺简单等优点的催化剂。
2 高效耐硫宽温Co-Mo基变换催化剂
高效耐硫Co-Mo基变换催化剂通常以Ⅵ和Ⅷ族中的某些金属(如Ni,Co,Mo,W 等)的氧化物或它们的混合物为活性组分,并添加碱金属(如K和Mg等)为功能性助剂。目前,工业上应用最多的是Co-Mo基宽温耐硫变换催化剂,在使用前将其转化为双金属硫化态Co-Mo,其中MoS2是催化剂的活性相,Co和 Ni是催化剂的助剂[33-34]。传统的Co-Mo基变换催化剂的制备方法有混碾法和浸渍法[35]。混碾法是将 Co、Mo(Ni,W)和碱金属的盐类水溶液、Al2O3粉、黏结剂等按规定配比混合,经碾压、造粒或压片、干燥、焙烧,得到催化剂成品(或将γ-Al2O3粉末浸渍上述盐类水溶液后挤条成型再经干燥焙烧而成)。浸渍法是将成型的γ-Al2O3等分步浸渍或共浸渍Co、Mo(Ni,W)和碱金属的盐类水溶液,经干燥、焙烧,得到催化剂成品。目前得到广泛应用的负载在γ-Al2O3上的Co-Mo-K/γ-Al2O3催化剂是一种性能优良的耐硫变换催化剂,既可作为中温耐硫变换催化剂使用,也可作为低温耐硫变换催化剂使用。
在研究和应用耐硫变换催化剂的过程中发现,以纯γ-Al2O3为载体的耐硫变换催化剂在高温、高压、高汽气比的条件下会出现γ-Al2O3的晶相变化、催化剂的活性和强度等指标大幅下降的缺点,进而又研究开发了复合载体,比较典型的载体有Mg-Al尖晶石、Ti-Al尖晶石、Ti改性的Mg-Al尖晶石等。在耐硫变换催化剂的Al2O3载体中加入MgO,特别是形成Mg-Al尖晶石的载体可以改善耐硫变换催化剂的强度、稳定性和宽温活性。Mg-Al尖晶石因在较高汽气比和压力下晶相不变、稳定性好和具有对耐硫变换有利的碱性等而被公认为是理想的耐硫变换催化剂载体[36-38]。特别是以Mg-Al尖晶石为载体的催化剂由于采用了特殊的制备工艺以及添加了新的组分和碱金属助剂,对高空速、宽汽气比的适应能力和稳定性高,具有较高的耐油、抗积碳和抗毒物的能力。因此Mg-Al尖晶石作为载体的重要作用越来越引起该领域科研人员的重视。
工业上通常可按是否含有碱金属K将催化剂分为Co-Mo-K基催化剂和无钾Co-Mo基催化剂,也可按照适用压力范围将催化剂分为两大类:一类是适用于低压(小于3.0 MPa)的耐硫低温变换催化剂Co-Mo-K/γ-Al2O3,这类催化剂制备工艺简单,低温活性高,但强度和稳定性差,存在K流失、易反硫化而失活的现象,主要在中小型合成氨厂的中低压流程中使用;另一类是适用于高压(3.0~8.0 MPa)、高汽气比(约1.4)的以Mg-Al尖晶石为载体的耐硫高温变换催化剂Co-Mo/MgO-Al2O3,这类催化剂具有较好的强度和稳定性,能在高压、高汽气比的大中型合成氨厂中的加压耐硫变换工艺中使用[30]。
国外在Co-Mo基耐硫变换催化剂的研制方面起步较早,目前已报道的耐硫变换催化剂的品种和型号较多,使用较多的工业耐硫变换催化剂[30-35]主要有:(1)1969年德国BASF公司开发成功的K8-11型耐硫变换催化剂,用于重油部分氧化法制合成气流程和加压煤气化制合成氨流程中的CO变换工序,首次在BASF公司路德维希氨厂使用。该型号催化剂的主要特点是以Mg-Al尖晶石为载体,硫化后活性高,耐高水蒸气分压,可在高压下使用,抗毒物能力强,能再生,平均寿命3~5 a;(2)由美国埃克森研究和工程实验室开发成功的SSK型催化剂,丹麦TopsΦe公司进一步开发,于1974年进行工业应用,主要用于重油氧化法CO变换工艺。该型号催化剂含有较高浓度的K2CO3促进剂,故低温活性高,同时对毒物不敏感,可耐100 μg/g的氯,存在K流失的缺点,尚无在7.84 MPa下使用的工业数据;(3)美国UCI公司开发的C25-2-02型新一代耐硫变换催化剂,主要用于低压流程。该型号催化剂的主要特点是含有稀土稳定剂和促进剂,催化剂的结构稳定性好,使用后强度和比表面积保留率高,低温活性好,抗毒能力强,但未见在7.4 MPa下使用的报道。
“十五”期间,国内建设了多套以煤或渣油为原料的大型合成氨厂,为配合引进装置所用耐硫变换催化剂的国产化以及满足国内中小型合成氨厂节能技术改造的需要,我国于20世纪80年代开展了耐硫变换催化剂的研制工作。上海化工研究院从1977年开始进行SB系列耐硫变换催化剂的研究,湖北省化学研究所重点研究了EB系列耐硫变换催化剂的制备及硫化方法,这两个系列催化剂属于Co-Mo-K/γ-Al2O3耐硫变换催化剂,主要用于中小型氮肥厂中温变换串联低温变换(简称中串低)或全程低温变换(简称全低变)工艺。中国石化齐鲁石化公司1988年开始耐硫变换催化剂的研制,并于1992年采用混合法开发出QCS系列的Co-Mo/MgO-Al2O3(或Co-Mo/MgO-TiO2-Al2O3)耐硫变换催化剂,主要用于由煤或渣油为原料高压气化生成的含硫原料气制取合成气和制氢的大型装置,目前生产的QCS-1型耐硫变换催化剂已部分替代BASF公司的K8-11型等国外催化剂,在高压水煤浆气化流程中应用。厦门大学于1998年开始开发新型水煤气变换催化剂和变换工艺。采用浸渍法制备了Co-Mo-W-K/γ-Al2O3和Co-Mo/MgO-Al2O3等XH系列多元组分变换催化剂及组合式填装方法,并于2006年在多家合成氨厂成功投用[39]。
国内外典型的Co-Mo基耐硫变换催化剂的性能及使用条件见表1。

表1 国内外典型的Co-Mo基耐硫变换催化剂的性能及使用条件Table 1 The properties of typical Co-Mo-based sulfur-tolerant water-gas shift catalyst at home and abroad
多年来,Co-Mo基变换催化剂在工业上不断得到推广应用,同时催化剂制备方法也不断改进,积累了许多宝贵的研究成果:
(1)载体与催化剂制备方法的改进[40-43]。采用模板剂或溶胶-凝胶法制备纳米粒子载体。相对于传统的混捏法,浸渍法可以提高活性组分在载体上的分散度,增加活性中心的数量并改善其分布。将多种活性组分一次负载到载体上的共浸渍法比传统分步浸渍法的浸渍次数少,催化剂活性高。在浸渍液中加入稳定剂,共浸渍后的物料经烘干和焙烧制得催化剂,使用时经硫化处理,其低温活性好。
(2)催化剂颗粒形态的优化。目前耐硫变换催化剂的颗粒形态一般为圆柱形或球形,若将催化剂制成其他形态,如三叶草和四叶草等,则可以增加颗粒外表面积和提高催化剂床层空隙率,从而提高单位体积催化剂的表观活性,降低床层流动阻力[44-45]。
(3)对载体进行改性或在催化剂中加入其他助剂[39,46-48]。采用拟薄水铝石为原料,通过添加造孔剂来调整载体的孔结构,并加入MgO提高载体强度。加入Ti促进催化剂低温下物理吸附H2S的能力。加入贵金属或稀土元素,可以使Mo保持较好的分散,提高催化剂的低温活性。在传统 Co-Mo-K/γ-Al2O3催化剂中,引入第三活性组分W,提高了催化剂的高温耐热性能。
3 Co-Mo基催化剂的催化反应机理
对于CO变换反应机理,最初人们认为催化剂的物理因素起着重要作用,后来在催化剂表面发现了CO与H2O的还原与氧化,提出了氧化-还原机理。随着研究的进一步深入,人们注意到催化剂表面处于微晶体角、棱、缺陷位置和不规则晶面上的原子和分子具有较大的不饱和性和最大的吸附能力,容易与反应物作用,从而确立了活性中心的概念并提出了吸附机理(缔合机理)。后来人们在催化剂上发现了甲酸中间物的形成,又提出了缔合机理-伴有中间物的吸附机理。为了更深入地阐明金属催化的水煤气反应机理,在羧甲基中间体机理的基础上又提出了双功能反应机理。CO和H2O的变换反应,按反应温度分为高温变换和低温变换,而两者的反应机理又有所不同[49-51]。通过对金属氧化物及金属催化生成的甲酸和甲酸根中间体的深入研究,通常认为在金属或碱性氧化物催化条件下,甲酸根中间体容易分解生成CO2和H2O,变换反应历程如下[52-53]:
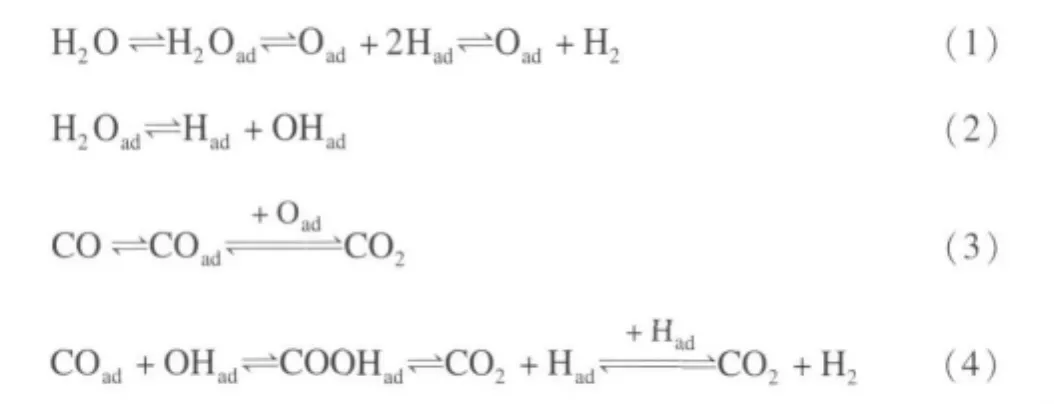
虽然近年来人们对水煤气变换反应进行了广泛而深入的研究,由于研究者采用的催化剂、实验条件、技术手段不尽相同,即使对同类型的机理,也可能会得到不同的表达式。CO变换反应或许不只以一种机理进行,更有可能是几种机理的综合作用,同时没有充足的证据说明在工业条件下哪种机理占优势。
对于Co-Mo基耐硫变换催化剂,尽管化学成分和含量不同,但主要活性组分是MoO3,且催化剂只有在硫化态下才具有变换活性。范淑蓉等[54]根据对Co-Mo-K/γ-Al2O3催化剂酸性的研究认为,变换反应与Mo5+无关,反应可能按Mo6+-Mo4+之间的双电子机理进行,而Mo4+为反应中心:

然而,Hou等[55]对MoO3基催化剂的变换反应动力学进行研究发现,原料气中的硫含量对反应活性起决定性的作用。根据动力学和催化剂的表征认为,在含硫水煤气变换反应中存在硫化和反硫化的动态平衡,在H2S/H2的还原气氛中,Mo6+离子还原成Mo5+和Mo4+的共存状态,变换反应是按照Mo5+-Mo4+之间的氧化-还原机理进行的:
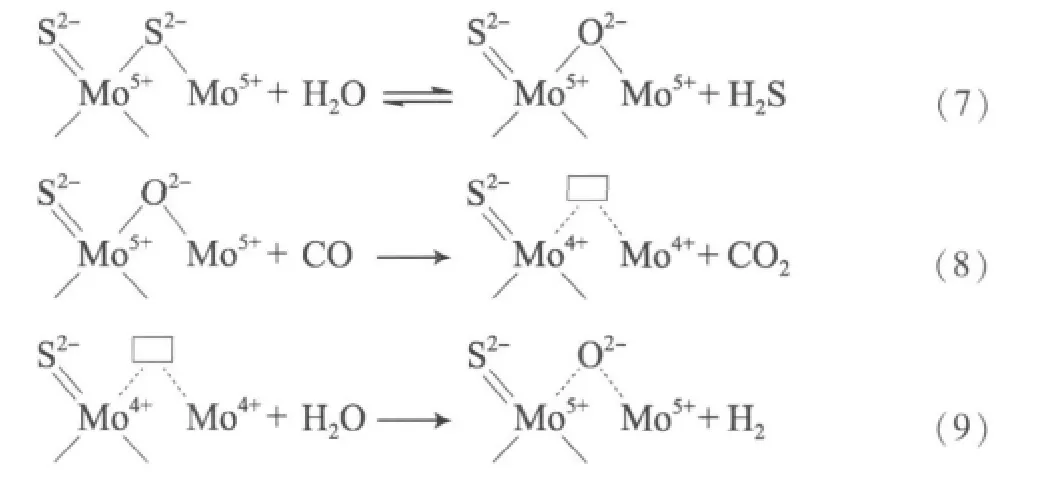
Nikolova等[56]认为在变换反应中,“thio-Mo5+”(S包围的 Mo)首先与水反应生成“oxysulfo-Mo5+”(O,S包围的 Mo),然后“oxysulfo-Mo5+”与CO反应生成 CO2,且 Mo5+还原为 Mo4+,然后Mo4+与水反应生成“oxysulfo-Mo5+”。随着对催化剂的活性与活性物种之间相互关系研究的深入,普遍认为硫化态Mo基催化剂在含硫原料气中的变换反应按照Mo5+-Mo4+之间的氧化-还原机理进行。同时从反应机理也可看出,由于反硫化反应的存在,会造成H2S的流失,因而在工业生产中原料气中要维持一定含量的H2S。
4 Co-Mo基催化剂的硫化及器外预硫化
工业上常用的Co-Mo基变换催化剂大多采用Mo,Co,Ni,W 等金属作为活性组分,并以氧化态形式分散在多孔载体上。Co-Mo基变换催化剂在使用前需要通过活化(硫化)的方法,使氧化态金属转变为具有催化活性的硫化态[57-58]。催化剂的硫化技术是催化剂开发和应用的关键步骤之一。传统的硫化方法基本上采用器内气相硫化工艺,即用原料气配入液体CS2高温氢解产生H2S,进行循环硫化或一次放空硫化;个别厂家采用高硫原料气直接硫化,也有采用固体硫化剂,即在反应器前串联一个硫化反应器,或将固体硫化剂与催化剂混合装填在加氢反应器内,通入还原气在器内实现硫化剂的热氢解反应产生H2S,H2S再与氧化态Co-Mo催化剂发生硫化反应。
器内气相硫化法的基本过程包括:催化剂的升温阶段(常温~220℃),主要是脱除催化剂中的物理水;催化剂的硫化阶段(220~350℃),主要是催化剂中表层的活性组分进行充分硫化;强化硫化阶段(350~420℃),主要通过升高硫化温度促使催化剂完全硫化;整个硫化时间一般在50 h以上。采用器内气相硫化法的优点在于可以减少硫化剂损失,硫化较为均匀,但存在以下缺点:需要专用的预硫化管路和设备;硫化时间较长;硫化过程中催化剂床层温度波动大;硫化剂均为有毒有害物质;易造成催化剂硫化不完全。在催化剂制备过程中,由于高温焙烧分解生成的氧化物与载体之间存在强烈的相互作用,往往导致硫化不完全[59-61]。
近年来,具有特有优点的器外预硫化技术得到普遍重视[59-61]。器外预硫化较常采用的方法是:将活性金属为氧化态的催化剂先与硫化剂混合,再装入反应器中,开工时只需通入氢气或同时通入含氢原料气,随后升温即可完成硫化。常用的硫化剂为单质硫、无机或有机多硫化物。器外预硫化技术的优点在于:开工时间短;活化期间催化剂床层温度稳定,容易操作;现场不需要再准备硫化剂,减少对环境的污染;不需要专用的预硫化设施;催化剂能够充分硫化;催化剂的活性较器内气相硫化的高或相当。目前器外预硫化技术在国外已经商业化,特别是在加氢脱硫工艺中;国内也进行了初步应用。因此,深入研究Co-Mo基催化剂的预硫化方法对开发高活性催化剂有重要意义。
Lian等[62]采用(NH4)2S溶液为预硫化剂制备了Co-Mo-S/MgO-Al2O3预硫化催化剂,研究发现,以H2S/H2/N2混合气进行器内气相硫化时,按照先硫化再还原的过程进行,也就是低温下先发生氧硫交换,MoO3前体中的端基O2-被S2-取代形成了Mo—S键,而氧硫交换继续进行,促使桥式S2-2和Mo5+中心形成;升高温度后Mo5+物种开始还原形成Mo4+存在于MoS2物种中。而在器外浸渍硫化铵的催化剂,先在催化剂表面形成含硫金属化合物前体,该含硫金属化合物前体在合成气条件下经历了热分解和还原过程[63-64]:(NH4)2MoS4(200 ~300℃)→MoS3(350~400℃)→MoS3(无定形态)(>500℃)→ MoS2(晶态)。器外预硫化与传统器内H2S硫化具有不同的活化机理。他们同时指出在预硫化时,Mo的化合物同样也会发生氧硫交换,在催化剂表面生成(NH4)2MoS4或(NH4)2MoOxSy,同时也可能存在部分其他硫化产物,如 MoOS,MoOS2,MoO2S等,甚至会出现组成和结构更为复杂的成分。在催化剂表面形成的硫化产物的表面上,硫是同时与Mo4+和Mo6+化合的,不仅仅存在于MoS2中。硫化态前体制备的MoS2/MgO-Al2O3催化剂本身已是硫化态,降低了活性组分与载体的相互作用,有利于形成 MoS2和CoMoS物种,提高硫化度,因而具有较高的变换活性和耐热性能。
先进的器外预硫化技术能克服传统器内气相硫化的缺点,使催化剂保持最佳的活性和稳定性,提高选择性,延长使用寿命。同时预硫化催化剂经过钝化处理后,可有效降低催化剂的自热性及SO2的生成,催化剂的固硫性能好,从而使催化剂保持活性和稳定性[65-66]。因此,深入研究CO变换催化剂的预硫化及钝化方法,对开发高活性、可工业化的预硫化CO变换催化剂有重大的现实意义。
5 高效节能变换工艺及其发展
煤气化是煤化工的龙头和基础,很大程度上影响煤化工的效率、成本和发展。根据原料种类和造气方法的不同,以煤为原料气的化工工艺可大致分为以下几种流程:常压固定层间歇气化、水煤浆加压气化、碎煤加压气化、粉煤加压气化。制取水煤气所用原料和气化工艺不同,水煤气中CO含量也不尽相同,例如,以煤或焦炭为原料,采用间歇式固定层煤气发生炉制得的水煤气中CO体积分数为25% ~30%;以天然气为原料,采用水蒸气转化法制得的水煤气中CO体积分数为13% ~18%;以重油或渣油为原料,经部分氧化制得的水煤气中CO体积分数为45%~50%;由水煤浆气化制得的半水煤气中CO体积分数为40% ~45%;粉煤加压气化制得的水煤气中CO体积分数高达60%。同时,不同的原料所制工艺气中硫的含量也有很大差异,因而选用的变换催化剂和变换工艺也有所不同[67-68]。然而,不管采用哪一种原料,合成氨生产工序都必须经过煤气化、水煤气变换、合成气净化、合成气压缩及合成过程,只不过在具体细节上有所不同,并随着合成氨技术的发展而不断演化[69]。
发达国家的变换工艺基本上都使用宽温型Co-Mo基催化剂取代传统的Fe-Cr基中温变换催化剂,而且国外的合成氨装置规模一般比较大,不管是原料、操作压力的选择、工艺流程还是催化剂与我国的中小型氮肥厂大不相同。我国氮肥企业开始于20世纪60年代,由常压变换改为加压变换,每吨氨水蒸气消耗高达1 200 kg;70年代高活性催化剂的普遍应用使每吨氨水蒸气消耗降到900 kg左右。随后在80年代我国开始引进国外第二代煤气化技术,采用加压连续气化制取合成氨原料气,气化压力既有较低的2.8 MPa,也有较高的6.5 MPa。加压连续气化制取的工艺气不仅含有大量的水蒸气,而且H2S和有机硫含量较高,其中,水蒸气可以用来进行变换反应,H2S可以保证Co-Mo基耐硫中温变换催化剂处于活性态,相应的CO变换反应大都要求采用加压耐硫变换工艺。Co-Mo基耐硫催化剂的研制成功使变换工艺得到重大变革,80年代由中温变换(简称中变)工艺改为中串低工艺,90年代发展了全低变工艺;为了克服全低变工艺不能长周期运行的问题,又开发了中温变换串联两段低温变换(简称中低低)工艺。
目前,国内中小型合成氨厂大都采用以煤为原料、间歇制气的生产工艺,与之配套的后续净化工艺主要有铜氨洗和甲烷化两种流程。铜氨洗净化流程要求变换气中CO体积分数低于3.0%,采用的变换工艺主要有中变工艺、中串低工艺、中低低工艺和全低变工艺等;而甲烷化净化流程则要求变换气中的CO体积分数小于0.3%,采用的变换工艺主要有两次脱碳的Fe-Cr基中变催化剂串联Cu-Zn基低温变换(简称低变)催化剂的变换工艺、Fe-Cr基中变催化剂串联二段Co-Mo基耐硫低变催化剂的中低低深度变换工艺、全部使用Co-Mo基耐硫低变催化剂的全低变深度变换工艺[70-72]。
5.1 与铜氨洗净化流程相配套的变换工艺
5.1.1 中变工艺
中变工艺一般是一个变换炉,炉内装填Fe-Cr基催化剂,分两段或三段装填,半水煤气从上到下依次通过各段催化剂完成变换过程。工艺指标:入炉半水煤气温度330~350℃、热点温度480~510℃、出炉变换气温度400~420℃、出口变换气中CO体积分数3% ~5%、入炉汽气比0.7~0.8、每吨氨水蒸气消耗1 000~1 200 kg。催化剂的终态温度较高,水蒸气消耗很大。目前采用中变工艺的合成氨厂逐渐减少。
5.1.2 中串低工艺
Fe-Cr基中变催化剂后再串联Co-Mo基耐硫低变催化剂,中变气经热交换后入低变炉。低变催化剂可放在中变炉最后一段,也可另设一低变炉串联在中变炉之后。中串低工艺流程的设置可分为炉内串低变增湿流程、炉内串低变调温水加流程、炉外中串低调温水加流程及炉外中串低增湿流程。工艺指标:中变炉入口温度320~340℃、热点温度460~480℃、中变炉出口温度380~400℃;低变炉入口温度200~220℃、低变炉出口温度240~260℃;中变炉出口CO体积分数5% ~6%、低变炉出口CO体积分数1% ~2%;入中变炉汽气比0.5~0.6、每吨氨水蒸气消耗500~600 kg。该工艺不仅低变催化剂的入口温度可降低100℃以上、每吨氨水蒸气消耗显著降低,取得了明显的节能效果,而且工艺改造较为简单,投资少,因此该工艺在低变工艺发展初期得到国内中小型合成氨厂的普遍采用。但是中串低流程存在明显的不足,主要是中变二、三段催化剂利用率很低,总的催化剂用量大,而生产能力却不能得到充分发挥。
5.1.3 中低低工艺
中低低变换工艺就是在中串低的基础上再串联一段低变段或低变炉。两个低变炉(段)之间要有降温措施,用水冷激或水加热器降温均可。一般有两种流程:中变增湿的中低低流程和调温水加的中低低流程。工艺指标:中变炉入口温度300~320℃、热点温度430~440℃、中变炉出口温度360~380℃;低变炉每段入口温度200~220℃、出口温度240~260℃;中变炉出口CO体积分数10% ~12%、低变炉出口CO体积分数1% ~2%、入炉汽气比0.4~0.5、每吨氨水蒸气消耗约400~500 kg。该工艺与中串低工艺本质上没有区别,由于多了低变炉(段),反应终态温度比中串低工艺低,因而汽气比有所下降,节能效果更佳,特别适合老变换系统扩大节能能力的改造。但中低低流程由于中变催化剂减少,一旦漏氧、热交换器或水加热器泄漏,第一低变催化剂极易中毒,因而要求催化剂有较强的抗毒性能,否则严重影响使用效果。同时由于第二低变段反应的汽气比、CO含量及反应温度都较低,要求第二低变段采用活性更高的催化剂,否则节汽效果不理想。
5.1.4 全低变工艺
全低变工艺是在中串低工艺基础上发展起来的全新变换工艺,变换炉各段采用Co-Mo基耐硫低变催化剂。全低变工艺的流程有两种:调温水加的全低变流程、增湿的全低变流程。工艺指标:入炉温度200~220℃、热点温度380~400℃左右、出炉温度250~260℃;变换炉出口CO体积分数1% ~2%、入炉汽气比0.3~0.4、每吨氨水蒸气消耗200~300 kg。相比而言,全低变工艺的节能效果最为明显,但全低变工艺反应温度低,难以分解煤气带入的焦油和润滑油,它们容易覆盖在催化剂表面,使一段Co-Mo基催化剂老化、失活,一段催化剂床层阻力容易升高,导致全低变工艺的工业应用成功率不高,未取得预期效果,相当一部分装置返回到中串低流程。
5.2 与甲烷化净化流程相配套的变换工艺
5.2.1 Fe-Cr基中变催化剂串联Cu-Zn基低变催化剂的变换工艺
Fe-Cr基中变催化剂工艺与铜氨洗净化流程中的Fe-Cr基中变催化剂工艺基本一致,但以煤为原料制半水煤气的工厂在Fe-Cr基中变催化剂后无法直接串联Cu-Zn基低变催化剂,需经二次脱碳、脱硫后再进入Cu-Zn基低变催化剂段。工艺指标:中变炉入口温度300~350℃、热点温度480~500℃、出口温度400~420℃、CO体积分数3% ~5%;低变炉入口温度190~200℃、出口温度230~250℃、出口CO体积分数小于0.3%;中变炉入炉汽气比0.8~0.9、低变炉入炉汽气比0.2~0.3、每吨氨水蒸气消耗1 100~1 300 kg。
5.2.2 中低低深度变换工艺
中低低深度变换工艺一般多采用二段Fe-Cr基中变催化剂串联二段Co-Mo基耐硫低变催化剂。工艺指标:中变炉入口温度300~320℃、热点温度460~480℃、出口温度380~400℃、出口CO体积分数5% ~6%;低变炉一段入口温度200~220℃、二段入口温度180~200℃、出口温度200~220℃、出口CO体积分数小于0.3%;中变炉入炉汽气比0.5~0.6、每吨氨水蒸气消耗约700~800 kg。
5.2.3 全低变深度变换工艺
全低变深度变换工艺与上述全低变工艺相似,一般采用三段Co-Mo基耐硫低变催化剂。工艺指标:一段入口温度200~220℃、热点温度380~400℃、二段入口温度200~220℃、三段入口温度180~200℃、出口温度210~230℃左右;变换出口CO体积分数小于0.3%、入炉汽气比0.5~0.6、每吨氨水蒸气消耗600~700 kg。
变换工艺的变革和进步始终围绕着催化剂、变换炉和变换气热能回收方式进行。变换反应中汽气比的高低直接影响变换工艺的水蒸气消耗,汽气比越低,过剩水蒸气量越小,因而全低变工艺的水蒸气消耗最低。同时在合成氨厂中,对变换气的高位能与潜热的综合利用对节能降耗起着重要的作用。早期,中变或中串低流程一般配置饱和热水塔进行热量回收,典型流程见图1。

图1 带饱和热水塔的中串低流程Fig.1 Simplified process flow diagram of medium-low temperature shift process with saturation tower.
随着低变技术的逐步成熟和完善,变换气中过量水蒸气已经很少,可以回收的热量也逐渐减少,因而开始采用无饱和热水塔变换工艺及副产中(低)压水蒸气技术,典型流程见图2。
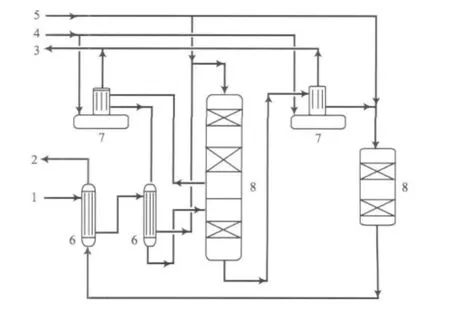
图2 无饱和热水塔的全低变流程Fig.2 Simplified process flow diagram of total low temperature shift process without saturation tower.
综合来说,变换工艺经历了常压变换到加压变换,在加压的基础上又经历了中变、中串低、中低低和全低变几个过程,每次技术革新,节能效果都明显改进。但并不是每一个工艺都完美无缺,它们都有其局限性,但总体来说节能效果越来越好。随着变换催化剂性能的不断改进以及新工艺和新设备的不断出现,将会促进热能的综合利用,变换系统的水蒸气消耗会大大降低。中变逐渐会被低变所代替,全低变技术将会成为CO变换工艺的主流方向。
6 结语
为了满足节能减排的需要,世界上出现了多种降低合成氨能耗的新工艺,特别是合成氨大型化、制气的加压化技术的发展,高活性低能耗变换催化剂及其催化工艺的开发越来越受到人们的重视,高效率节能型变换工艺将是今后变换工艺的发展方向。
变换工艺的核心在于催化剂,催化剂的性能直接影响到变换工艺的效能。通过综合分析中变、中串低、中低低和全低变4种代表性变换工艺及其催化剂的特点可知,目前在中小型合成氨厂能稳定运行的流程为中低低变换工艺,节能效果最佳的流程为全低变工艺,但其所用一段全低变催化剂的性能尚需改进。因此开发出能适应中低压变换反应炉一段变换的耐硫变换催化剂,对我国中小型合成氨厂进行节能改造具重要意义。同时深入研究CO变换催化剂的预硫化及其钝化方法,开发高活性、可工业化的预硫化CO变换催化剂将是另一重要课题。
[1]Antonio A,Sigurd S.Control Structure Design for the Ammonia Synthesis Process[J].Comput Chem Eng,2008,32(12):2920-2932.
[2]Deng Xue,Wang Hewu,Huang Haiyan,et al.Hydrogen Flow Chart in China[J].Int J Hydrogen Energy,2010,35(13):6475-6481.
[3]Longwell J P,Rubin E S,Wilson J.Coal:Energy for the Future[J].Progr Energ Combust Sci,1995,21(4):269-360.
[4]於子方.90年代合成氨工艺技术的新进展[J].石油化工动态,1997,5(5):33-38.
[5]陈元春,金小娟.我国煤化工产业发展状况评述[J].煤炭工程,2009,(5):90-92.
[6]Dong Ju Moon.Low Temperature WGS Catalysts for Hydrogen Station and Fuel Processor Applications[J].Catal Surv Asia,2009,13(3):191-204.
[7]李速延,周晓奇.CO变换催化剂的研究进展[J].煤化工,
2007,129(2):31-34.
[8]Ruettinger W,Ilinich O,Farrauto R J.A New Generation of Water Gas Shift Catalysts for Fuel Cell Applications[J].J Power Sources,2003,118(1-2):61-65.
[9]Galvita V,Schröder T,Munder B,et al.Production of Hydrogen with Low COx-Content for PEM Fuel Cells by Cyclic Water Gas Shift Reactor[J].Int J Hydrogen Energy,2008,33(4):1354-1360.
[10]Laniecki M,Malecka-Grycz M,Domka F.Water-Gas Shift Reaction over Sulfided Molybdenum Catalysts:Ⅰ.Alumina Titania and Zirconia-Supported Catalysts[J].Appl Catal,A,2000,196(2):293-303.
[11]Baier T,Kolb G.Temperature Control of the Water Gas Shift Reaction in Microstructured Reactors[J].Chem Eng Sci,2007,62(17):4602-4611.
[12]杨玲菲,宁平,田森林,等.低汽气比节能变换催化剂研究进展[J].化工进展,2009,28(S1):49-53.
[13]Penã M A,Gómez J P,Fierro J L G.New Catalytic Routes for Syngas and Hydrogen Production[J].Appl Catal,A,1996,144(1-2):7-57.
[14]Liu Quansheng,Ma Wenping,He Runxia,et al.Reaction and Characterization Studies of an Industrial Cr-Free Iron-Based Catalyst for High-Temperature Water Gas Shift Reaction[J].Catal Today,2005,106(1-4):52-56.
[15]Martos C,Dufour J,Ruiz A.Synthesis of Fe3O4-Based Catalysts for the High-Temperature Water Gas Shift Reaction[J].Int J Hydrogen Energy,2009,34(10):4475-4481.
[16]吕待清.铁基高温变换催化剂热力学稳定性分析[J].化肥工业,2006,33(5):25-29.
[17]欧晓佳,程极源.铁铬系高(中)温变换催化剂的研究现状[J]. 化学研究与应用,1999,11(2):126-131.
[18]Lei Yun,Cant N W,Trimm D L.The Origin of Rhodium Promotion of Fe3O4-Cr2O3Catalysts for the High-Temperature Water-Gas Shift Reaction[J].J Catal,2006,239(1):227-236.
[19]华南平,杨平,杜玉扣.CO高温变换催化剂发展趋势[J].小氮肥设计技术,2005,26(4):1-4.
[20]Lee Joon Yeob,Lee Dae-Won,Lee Kwan-Young,et al.Cr-Free Fe-Based Metal Oxide Catalysts for High Temperature Water Gas Shift Reaction of Fuel Processor Using LPG[J].Catal Today,2009,146(1-2):260-264.
[21]Gawade P,Mirkelamoglu B,Tan B,et al.Cr-Free Fe-Based Water-Gas Shift Catalysts Prepared Through Propylene Oxide-Assisted Sol-Gel Technique[J].J Mol Catal A:Chem,2010,321(1-2):61-70.
[22]Lima A A G,Nele M,Moreno E L,et al.Composition Effects on the Activity of Cu-Zn-Al Based Catalysts for the Water Gas Shift Reaction:A Statistical Approach[J].Appl Catal,A,1998,171(1):31-43.
[23]李速延,周晓奇.CO变换催化剂的研究进展[J].煤化工,2007,129(2):31-34.
[24]Wu Jingang,Saito Masahiro.Improvement of Stability of a Cu/Zn/Al Catalyst for the CO Shift Reaction[J].J Catal,2000,195(2):420-422.
[25]李选志,韦孙昌.铜基一氧化碳低温变换催化剂的研究进展[J].大氮肥,2006,29(1):69-72.
[26]Koryabkina N A,Phatak A A,Ruettinger W F,et al.Determination of Kinetic Parameters for the Water-Gas Shift Reaction on Copper Catalysts Under Realistic Conditions for Fuel Cell Applications[J].J Catal,2003,217(1) :233-239.
[27]Yahiro Hidenori,Nakaya Kenta,Yamamoto Tetsuya,et al.Effect of Calcination Temperature on the Catalytic Activity of Copper Supported on γ-Alumina for the Water-Gas-Shift Reaction[J].Catal Commun,2006,7(4):228-231.
[28]Panagiotopoulou P,Kondarides D I.A Comparative Study of the Water-Gas Shift Activity of Pt Catalysts Supported on Single(MOx)and Composite(MOx/Al2O3,MOx/TiO2)Metal Oxide Carriers[J].Catal Today,2007,127(2)319-329.
[29]刘全生,张前程,马文平,等.变换催化剂研究进展[J].化学进展,2005,17(3):389-398.
[30]Nagai Masatoshi,Matsuda Kenji.Low-Temperature Water-Gas Shift Reaction over Cobalt-Molybdenum Carbide Catalyst[J].J Catal,2006,238(2):489-496.
[31]Laniecki M,Ignacik M.Water-Gas Shift Reaction over Sulfided Molybdenum Catalysts Supported on TiO2-ZrO2Mixed Oxides Support Characterization and Catalytic Activity[J].Catal Today,2006,116(2):400-407.
[32]Chianelli R R,Berhault G,Torres B.Unsupported Transition Metal Sulfide Catalysts:100 Years of Science and Application[J].Catal Today,2009,147(3-4) :275-286.
[33]Hakkarainen R,Salmi T,Keiski R L.Water-Gas Shift Reaction on a Cobalt-Molybdenum Oxide Catalyst[J].Appl Catal,A,1993,99(2) :195-215.
[34]Mohamed M M,Salama T M,Othman A I,et al.Low Temperature Water-Gas Shift Reaction on Cerium Containing Mordenites Prepared by Different Methods[J].Appl Catal,A,2005,279(1-2):23-33.
[35]石自更,王峰.K8-11和QDB-04型耐硫变换催化剂在壳牌粉煤气化制甲醇装置的应用[J].化肥设计,,2009,47(2):33-36.
[36]Kirszensztejn P,Przekop R,Szymkowiak A,et al.Preparation of MgO-Al2O3Binary Gel System with Mesoporous Structure[J].Microporous Mesoporous Mater,2006,89(1-3):150-157.
[37]袁素珺,张青红,李耀刚,等.高比表面积的镁铝复合氧化物纳米薄片的制备及其吸附性能[J].硅酸盐通报,2009,28(4):636-640.
[38]Sarkar R,Bannerjee G.Effect of Addition of TiO2on Reaction Sintered MgO-Al2O3Spinels[J].J Eur Ceram Soc,2000,20(12):2133-2141.
[39]Wang Huifang,Lian Yixin,Li Yinong,et al.W-Promoted Co-Mo-K/γ-Al2O3Catalysts for Water-Gas Shift Reaction[J].Catal Commun,2009,10(14):1864-1867.
[40]李小定,陈劲松.钴钼系一氧化碳变换催化剂的制备方法:中国,90102336[P].1993-10-27.
[41]Nickolov R N,Edreva-Kardjieva R M,Kafedjiysky V J,et al.Effect of the Order of Potassium Introduction on the Texture and Activity of Mo/Al2O3Catalysts in Water Gas Shift Reaction[J].Appl Catal,A,2000,190(1):191-196.
[42]Nagai M,Zahidul Md A,Matsuda K.Nano-Structured Nickel-Molybdenum Carbide Catalyst for Low-Temperature Water-Gas Shift Reaction[J].Appl Catal,A,2006,313(2):137-145.
[43]连奕新,王会芳,张元华,等.焙烧温度对镁铝复合氧化物载体性能的影响[J]. 石油化工,2009,38(6):622-628.
[44]纵秋云,田兆明,谭永放,等.QCS-4耐硫变换催化剂的研制[J]. 齐鲁石油化工,1998,26(1):14-18.
[45]Mellor J R,Coville N J,Sofianos A C,et al.Raney Copper Catalysts for the Water-Gas Shift Reaction:Ⅰ.Preparation,Activity and Stability[J].Appl Catal,A,1997,164(1-2):171-183.
[46]Nagai M,Zahidul Md A,Kunisaki Y,et al.Water-Gas Shift Reactions on Potassium-and Zirconium-Promoted Cobalt Molybdenum Carbide Catalysts[J].Appl Catal,A,2010,383(1-2):58-65.
[47]Panagiotopoulou P,Kondarides D I.Effect of Morphological Characteristics of TiO2-Supported Noble Metal Catalysts on Their Activity for the Water-Gas Shift Reaction[J].J Catal,2004,225(2):327-336.
[48]Boccuzzi F,Chiorino A,Manzoli M,et al.Gold,Silver and Copper Catalysts Supported on TiO2for Pure Hydrogen Production[J].Catal Today,2002,75(1):169-175.
[49]郭晓勇.负载型金属纳米催化剂的制备及其对水煤气的催化变换作用[D].苏州:苏州大学,2007.
[50]Rhodes C,Hutchings G J,Ward A M.Water-Gas Shift Reaction:Finding the Mechanistic Boundary[J].Catal Today,1995,23(1):43-58.
[51]Natesakhawat S, Wang Xueqin, Zhang Lingzhi, etal.Development of Chromium-Free Iron-Based Catalysts for High-Temperature Water-Gas Shift Reaction[J].J Mol Catal A:Chem,2006,260(1-2):82-94.
[52]Shi Xuerong,Wang Shengguang,Hua Jia,et al.Density Functional Theory Study on Water-Gas-Shift Reaction over Molybdenum Disulfide[J].Appl Catal,A,2009,365(1):62-70.
[53]Li Yumin,Wang Rejie,Chang Liu.Study of Reactions over Sulfide Catalysts in CO-CO2-H2-H2O System[J].Catal Today,1999,51(1):25-38.
[54]范淑蓉,谢筱帆,窦伯生,等.水煤气变换催化剂Co-Mo-K/γ-Al2O3的酸性对活性的影响[J].应用化学,1991,8(3):42-45.
[55]Hou P,Meeker D,Wise H.Kinetic Studies with a Sulfur-Tolerant Water Gas Shift Catalyst[J].J Catal,1983,80(2):280-285.
[56]Nikolova D,Edreva-Kardjieva R,Gouliev G,et al.The State of(K)(Ni)Mo/γ-Al2O3Catalysts After Water-Gas Shift Reaction in the Presence of Sulfur in the Feed:XPS and EPR Study[J].Appl Catal,A,2006,297(1):135-144.
[57]Eijsbouts S,Mayo S W,Fujita K.Unsupported Transition Metal Sulfide Catalysts:From Fundamentals to Industrial Application[J].Appl Catal,A,2007,322(1):58-66.
[58]Frizi N,Blanchard P,Payen E,et al.Genesis of New HDS Catalysts Through a Careful Control of the Sulfidation of Both Co and Mo Atoms:Study of Their Activation Under Gas Phase[J].Catal Today,2008,130(2-4):272-282.
[59]Frizi N,Blanchard P,Payen E,et al.Genesis of New Gas Oil HDS Catalysts:Study of Their Liquid Phase Sulfidation[J].Catal Today,2008,130(1):32-40.
[60]王月霞.加氢催化剂的器外预硫化[J].炼油设计,2000,30(7):57-58.
[61]van Gesetl J,Leglise J,Duehet J C.Catalytic Properties of a CoMo/Al2O3Catalyst Pre-Sulfided with Alkylpoyl Sulfides:Comparison with Conventional Sulfiding[J].J Catal,1994,145(2):429-436.
[62]Lian Yixin,Wang Huifang,Fang Weiping,et al.Water Gas Shift Activity of Co-Mo/MgO-Al2O3Catalysts Presulfided with Ammonium Sulfide[J].J Nat Gas Chem,2010,19(1):61-66.
[63]Yoosuk Boonyawan,Kim Jae Hyung,Song Chunshan,et al.Highly Active MoS,CoMoS and NiMoS Unsupported Catalysts Prepared by Hydrothermal Synthesis for Hydrodesulfurization of 4,6-Dimethyldibenzothiophene[J].Catal Today,2008,130(1):14-23.
[64]柴永明,赵会吉,柳云骐,等.四硫代钼酸铵制备方法的改进[J]. 无机盐工业,2007,39(5):12-15.
[65]高善彬,董群,刘滨,等.预硫化加氢催化剂钝化机理的研究进展[J].化学工业与工程,2008,25(2):183-188.
[66]王宏悦,潘兆德.器外预硫化耐硫变换催化剂的研制[J].煤化工,2009,140(1):18-20.
[67]杨余芳.一氧化碳加压变换系统的探讨与设计[D].湘潭:湘潭大学,2002.
[68]Heidebrecht P,Sundmacher K.Thermodynamic Analysis of a Cyclic Water Gas-Shift Reactor(CWGSR)for Hydrogen Production[J].Chem Eng Sci,2009,64(23):5057-5065.
[69]王明华,李政,倪维斗.煤制甲醇CO变换工艺组合方式的研究[J].化学工程,2008,36(7):66-70.
[70]侯引平.中小型合成氨装置的变换工艺[D].天津:天津大学,2005.
[71]Chen Wei-Hsin,Lin Mu-Rong,Jiang Tsung Leo,et al.Modeling and Simulation of Hydrogen Generation from High-Temperature and Low-Temperature Water Gas Shift Reactions[J].Int J Hydrogen Energy,2008,33(22):6644-6656.
[72]Levent M.Water-Gas Shift Reaction over Porous Catalyst:Temperature and Reactant Concentration Distribution[J].Int J Hydrogen Energy,2001,26(6):551-558.
Co-Mo-Based Catalyst and Catalytic Reaction Process for Water-Gas Shift
Lian Yixin,Yang Yiquan,Fang Weiping
(College of Chemistry and Chemical Engineering,National Engineering Laboratory for Green Chemical Productions of Alcohols,Ethers and Esters,Xiamen University,Xiamen Fujian 361005,China)
The development background,present situation and developing vista for CO shift catalysts,and the process technologies for water-gas shift reaction both at home and abroad were introduced.The mechanism of the water-gas shift reaction and the characteristic of the catalyst sulfuration methods were also expounded.By comparing the industrial applications of the catalysts in four cross-sectional shift processes,i.e.the high temperature shift,the high-low temperature shift,the high-low-low temperature shift and the total low temperature shift,it can be imagined that to investigate and develop the Co-Mo-based shift catalysts with high activity and stability,which suit the total low-temperature shift process with energy saving,and the preparation procedures for the catalyst pre-sulfuration will be the developing direction from now on.
water-gas shift;cobalt-molybdenum-based catalyst;total low temperature shift;carbon monoxide;hydrogen;carbon dioxide
1000-8144(2011)04-0347-11
TQ 546.4
A
2010-09-19;[修改稿日期]2010-12-25。
连奕新(1971—),男,福建省惠安县人,博士,工程师,电邮lianyx@xmu.edu.cn。联系人:方维平,电话0592-2186291,电邮wpfang@xmu.edu.cn。
本文从水煤气变换催化剂的现状与发展出发,着重介绍了目前两类高效耐硫Co-Mo基变换催化剂的应用与研究情况,阐述了Co-Mo基催化剂的催化机理与硫化方法,进而对比了不同变换工艺流程的操作与能耗,指出适合节能型工艺的Co-Mo基耐硫变换催化剂及其预硫化技术是今后的研究方向。
(编辑 王 萍)
猜你喜欢
中国化肥信息(2022年4期)2023-01-02
中氮肥(2021年2期)2021-12-25
中氮肥(2021年5期)2021-12-23
中国特种设备安全(2019年3期)2019-04-22
石油化工自动化(2018年5期)2018-11-14
山东工业技术(2016年15期)2016-12-01
山东工业技术(2016年15期)2016-12-01
安徽化工(2016年4期)2016-11-29
现代盐化工(2016年1期)2016-05-23
环境科技(2015年5期)2015-11-08
