
聚硅酸制备的数学建模及聚铁混凝剂性能
2010-11-16 08:08于水利杨园晶
哈尔滨工业大学学报 2010年2期
付 英,于水利,杨园晶
(1.哈尔滨工业大学 市政环境工程学院,哈尔滨150090,fuying8899@163.com;2.济南大学 土木建筑学院,济南250022)
由于铝在生物体内具有累积毒性[1],对硅-金属盐类[2-3]混凝剂的研究由聚硅酸铝转向聚硅酸铁[4].作为制备聚硅酸铁(PSF)混凝剂的主体之一,聚硅酸(PS)的制备方法是Baylis[5]在1937年首次提出的.合成PS 的一个基本指标是聚合度(聚合时间/凝胶时间),聚合度是影响PSF 物化特征、微观形态及混凝性能的重要因子.影响聚合度的主要因素有PS 的聚合pH 值、w(SiO2)及温度等.一般来讲,实验室是在一些固定合成PS 条件下研制PSF,成功的PSF 配方具有一些特定条件,尤其是PS 的合成条件,比如固定的pH、温度等,而从成本考虑,聚合温度一般都控制在室温左右.要把PSF 真正工业化,就可能由于一些实际情况的变化导致PSF 配方品质的下降,比如现场的环境条件、生产设施、冬夏温度及原料特性等都会有较大差异.为使科研活动与产品工业化接轨,针对影响PS 聚合度的主要因素,对PS 的合成过程进行数学建模,可以在多种条件下根据所需要的聚合度计算聚合时间.本文采用共聚法制备PSF,并对比研究PSF、PFS 及PFA 对低温低浊水的混凝性能及其微观品质,为PSF 的研制及应用提供基础数据.
1 实 验
1.1 主要仪器和试剂
SPECTRU 1 红外光谱仪(美国),JEM-1200EX 型透射电子显微镜(TEM,日本),ZS 纳米粒度分析仪(英国),AQ2010 浊度仪(美国),DHSJ-3F 型pH 计(上海),MY3000-6K 型自动控制六联搅拌仪(湖北),T6 新世纪紫外/可见分光光度计(北京).水玻璃(w(SiO2)=26%,模数为3.16,密度为1.36 kg/L)、FeSO4·7H2O、Na-ClO3、H2SO4均为工业级原料,PFS、PFA 为某水厂提供.实验用水为重蒸水.
1.2 PS 的制备及数学建模
采用酸性条件制备PS.将水玻璃稀释为3%~13%,在高速搅拌条件下缓慢加入到稀H2SO4溶液(20%)中,控制pH 值为1.5 ~4.0,温度为2~40 ℃,观察挂壁及成冻时间.用该时间对制备条件做图,然后进行曲线拟合,建立数学模型.
1.3 PSF 的制备
将水玻璃稀释为3.5%~9.0%,在高速搅拌条件下缓慢加入到20%的H2SO4溶液中,控制pH 值为1.5 ~3.5,室温下聚合1 ~5 h,制得半成品PS.根据不同的Si/Fe 比,将10.52 ~35.21 g FeSO4·7H2O 溶解到稀H2SO4溶液中,然后在40~60 ℃下与PS 快速混合,同时加入0.32 ~0.90 g NaClO3,熟化,加入稳定剂,稀释,制得不同Si/Fe 比的PSF 样品,Si/Fe 摩尔比分别为0.5,1,3,以PSF0.5、PSF1 及PSF3 表示,c(总Fe)=0.18 mol/L.
1.4 混凝实验
实验用水取自污染严重的冬季松花江水,自然沉降24 h 后:浊度为5.4 ~6.7 NTU,温度为5 ℃,pH 为7.17,UV254=0.116 ~0.134 cm-1,CODMn=4.32 ~5.14 mg/L(以O2计),投药浓度为0.018 mol/L,有效成份与下面投药量一样,PSF、PFS 均以Fe 计,PFA 以Al 计.固定混凝剂投量,将1 L 水样置于搅拌机上,快速搅拌瞬间投加混凝剂.搅拌程序为:快搅200 r/min,1 min;慢搅40 r/min,10 min;沉降不同时间,在液面下2 cm处取300 mL 上清液测定浊度、pH、UV254及CODMn.
1.5 混凝剂微观品质的表征
微观形态:吸取少量的液体PSF1、PFS 及PFA,滴到带有支持膜的铜网上,干燥数分钟后,置于TEM 下观察拍照.
Zeta 电位及粒径:用Zeta 电位仪分别测定上述3 种混凝剂的Zeta 电位及粒径,测定次数为6~10 次.
红外光谱:将液体混凝剂置于50 ℃的烘箱中干燥制成粉末状固体,采用KBr 压片法绘制红外光谱图.
2 结果与讨论
2.1 PS 的凝胶时间、pH 变化规律及数学建模
具有一定聚合度的PS 是制备PSF 的半成品原料,Fe3+具有很强的水解倾向,碱性条件合成的PS 不适合做PSF 的原料,本文关于PS 的一切研究都是在酸性条件下进行的,具有实际价值.
2.1.1 PS 凝胶时间及pH 变化规律
1)PS 凝胶时间与制备条件的关系
图1(a)中,w(SiO2)=6%,温度为25 ℃;图1(b)中,pH=3,温度为25 ℃;图1(c)中,w(SiO2)=6%,pH=3.
本实验中w(SiO2)相对较高,各因素对凝胶时间的影响与低w(SiO2)的情况[6]有很大不同,主要表现在制备条件不同区间的斜率及曲线走势的差异.由图1(a)看出,随pH 的增加,PS 的凝胶时间倾向于直线快速下降.图1(b)中,凝胶时间与w(SiO2)的关系有一个突变,w(SiO2)<6%时,凝胶时间降低很快,之后下降缓慢,假设按直线拟合,两段的斜率分别为-14.99,-0.895,突变值的出现说明在不同初始w(SiO2)下,可能出现硅酸各形态的析出速度、饱和浓度以及三者间的比例等有很大差异,导致PS 可能具有不同的聚合反应机理,这方面值得进一步探讨.图1(c)出现两个拐点,2 ~5 ℃、15 ~25 ℃时凝胶时间受温度影响最大,5 ~15 ℃之间次之,而25 ~40 ℃区间内随温度变化缓慢.对于挂壁与成冻的关系来说,pH、w(SiO2)曲线都是随其值的增加而间隔减小,而对于温度来说,凝胶时间受温度影响较大处其间隔较小,这些对于PS 作为助凝剂是很有用的信息.

图1 PS 制备条件对其凝胶时间的影响
2)聚合温度对PS 聚合过程中pH 值的影响
如图2 所示,w(SiO2)=6%,pH=3.20 ℃时PS 的凝胶时间较长,达到19 h,因此,2.5 h 之内pH 的变化很小.随温度的升高聚合初期pH 下降,然后上升,原理与戴安邦硅酸聚合的酸性机制相吻合,主要是H4SiO4与H5SiO4+六配位的羟连作用[7],该反应过程中并不释放OH-,PS 的酸离常数比单硅酸大[8],且随聚合度的升高而增加,释放质子的能力增强,因此,溶液的pH 逐渐降低.然后由于多硅酸的酸离解而又带负电荷,带负电的多硅酸与H4SiO4进行释放OH-离子的反应[7],使溶液pH 升高.由图2 看出,温度越高,pH下降或上升的速度越快,实验中还发现,40,50,60 ℃的凝胶时间分别为2.75,1,0.67 h,凝胶时pH 值分别为2.55,2.46,2.39,可以这样解释,温度越高,电子、质子越活跃,从而越容易脱离母体分子而与临近分子结合,这样与Si 相连的6 个键端OH-离子可以与临近硅酸分子同时发生连锁反应,速度急剧增加,因此,pH 变化速度快,凝胶时间随温度升高而缩短.
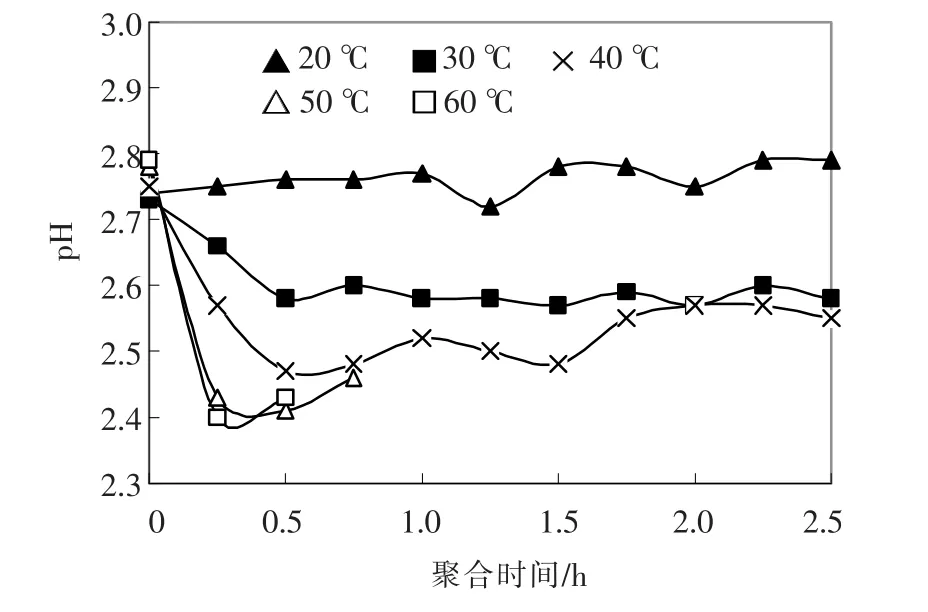
图2 温度对PS 聚合过程pH 值的影响
3)PS 聚合反应中改变pH 值前、后的pH 变化
w(SiO2)=6%,pH=3,温度为25 ℃.将聚合5 h 的PS 取出一半,将pH 值调到2,并同时监测改变pH 前(PS1)、后(PS2)两个样品的pH 变化情况,如图3 所示.随聚合时间的增加,PS1 的pH先略微降低,然后上升,但变化很小.PS2 的pH 上升速度很快,然后趋于定值,这是因为调低pH 时PS 已经达到一定聚合度,处于释放OH-离子的反应阶段,pH 降低导致带负电的PS 质子化,中性H4SiO4含量增加,反应速度加快,释放的OH-离子增多,表现为pH 上升快.
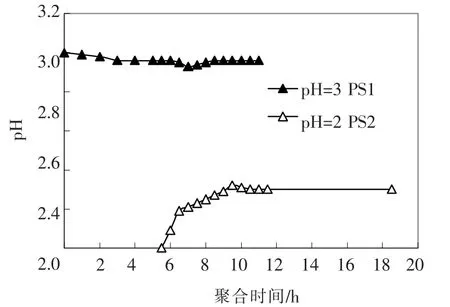
图3 改变pH 对具有一定聚合度PS pH 值的影响
由图1(a)看出,初始pH 值为2、3 的凝胶时间分别为33,12.5 h,而将pH=3、聚合度为40%(聚合5 h)的PS 的pH 值调低到pH=2 后(图3),其凝胶时间为18.5 h.由极限黏度与聚合度的关系可知[9],PS 聚合过程并不是均匀过程,聚合度为70%以前较均匀,之后聚合速度加快,因此,可以认为本实验中PS(聚合度为40%)是均匀聚合过程.根据不同pH 值时PS 的聚合度换算可知,将pH 调低后其凝胶时间为26.7 h,即改变pH 后的PS 其凝胶时间缩短,说明将PS 聚合到一定程度后再调低pH 值,部分PS 将发生解聚,但是解聚后的状态完全不同于初始同样低pH 值的状态,这可能是采用酸化剂来延缓PS 聚合过程的本质原因.
2.1.2 PS 凝胶时间的数学建模
这里t 指成冻时间.本文采取多项式建模,图1 中散点的数学多项式模型如表1 所示.为提高拟合精度,t-w(SiO2)、t-温度采取分段拟合的方式.在t-温度图中,由于40 ℃时的回归值与测定值误差太大,建模时略去该点.

表1 PS 凝胶时间的数学模型表
表1 中y 代表凝胶时间,x 代表制备条件.R2表示相关系数,作为衡量配后曲线效果好坏的指标.残余平方和Q 作为回归方程预报Y 值的精度指标.经过计算,对于pH、w(SiO2)及温度,Q 分别为0.011 7,7.383 4 及19.202,残余标准差σ 分别为0.054 1,1.109 3 及1.656 2.综合Q、R2及σ看出,t-pH 曲线拟合的效果最好,Q、σ 最小,R2最大,而t-温度曲线的拟合精度相对较差.由表2看出,较大的误差都出现在曲线拐点处,这是多项式建模的缺陷.
PS 聚合是相当复杂的反应过程,可能是多种机制并存,又由于该过程不是均匀过程,不同条件下其不均匀程度完全不同,工业化生产PSF 时,如果只根据经验数据来定性确定不同现场环境下的PS 聚合时间,必定导致PSF 配方品质的下降.因此,对PS 的制备过程进行多项式分段数学建模是工业制备优质PSF 的基础条件,这样就可以在多种条件下根据所需要的聚合度计算聚合时间,使PS 的制备更具有环境缓冲能力,使PSF 的工业化产品更具有优异的性能.期待对这方面进行更多的研究及出现更精确的模型.
2.2 高分子铁类混凝剂处理低温低浊水性能对比
从图4 看出,PSF 的混凝效果明显优于PFS、PFA.投量低时,PSF3 去除UV254的性能较好,但随投量的增加其优势逐渐降低,因为对溶解性有机物主要是通过电中和/脱稳、共沉淀去除,而PSF3由于PS 含量高导致总正电荷较低,所以,随投量的增加,正电荷较高的PSF0.5、PSF1逐渐占有优势.PSF1 对UV254及CODMn的去除率比PFS、PFA分别约高出10%及17%、40%及20%多.随投药量增加,PSF 的沉后水pH 值下降较多,而PFS、PFA 比较平稳,这是PSF 的缺陷之一,但可以满足饮用水的要求.
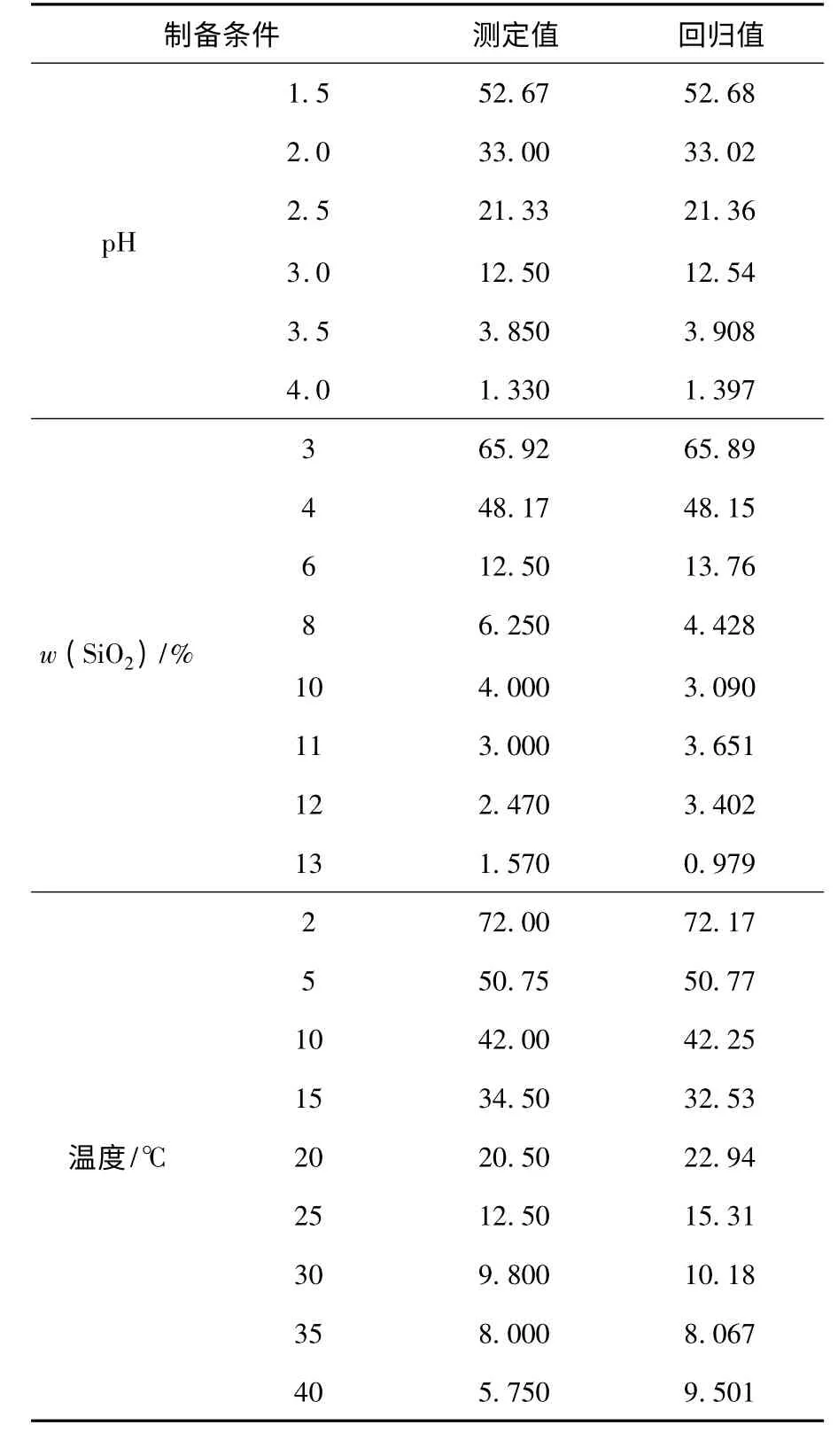
表2 凝胶时间的测定值与回归值的对比 h
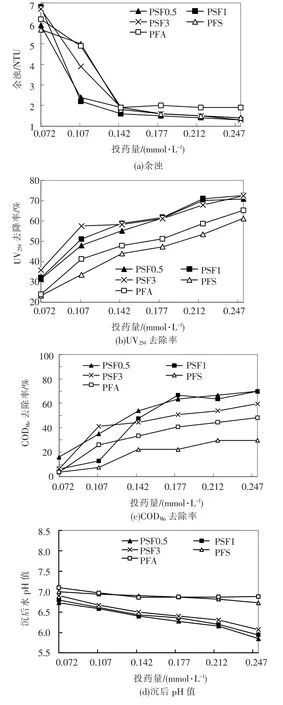
图4 投药量对混凝性能及沉后水pH 值的影响
2.3 高分子铁类混凝剂的微观品质对比
2.3.1 微观形态、Zeta 电位及粒径特征
由图5(a)看出,PSF1 是由许多链节样物种连接而成的分维数很大的敞开式枝状结构,并且形态大小覆盖范围很宽,这与粒径测定结果吻合.PFS 与PFA 是由一些低分维数及尺寸很小的无规形态组成(图5(b)、5(c)),其中PFS 有团聚倾向,而PFA 分布比较松散.PSF1、PFS、PFA 的Zeta电位平均值分别为1.75,1.92,36.1 mV,平均粒径分别为2 965,629.7,267.4 nm.

图5 混凝剂电镜图片
2.3.2 红外光谱特征
由图6 看出,3 188 ~3 522 cm-1处的峰是表示与Fe 或Al 相连或游离的—OH 的伸缩振动吸收峰[9],与PSF1 相比,PFS 中的该峰分裂为两个峰3 522 及3 188 cm-1,说明有振动的偶合现象存在[10],可能是Fe—OH 键互相交叉连接的结果,是多核羟基桥连的证明.1 227 ~1 380 cm-1表示与Fe 相连的—OH 的伸缩振动峰,与PFS 相比,PSF1中的该峰峰位发生红移,这是空间位阻效应[10]的结果,说明PSF 空间结构连接紧密,是枝杈结构的特征.974 cm-1处的峰位归属为Si—O—Fe 键[11],这是PSF1 独有的特征峰,说明PSF 是Fe 与Si 的共聚产物,而不是简单的原料混合.反映Fe—OH—Fe[12]的特征峰是725 ~884 cm-1,在PFS中,725 cm-1处的强宽吸收峰可能是以羟基桥连的多核Fe—OH—Fe 的伸缩振动吸收峰,在PFS中有比在PSF1 中较低的—OH 键频率,说明PSF1中的—OH 主要是分子内氢键[13],而PFS 分子间氢较多,这是PSF1 为枝杈结构、而PFS 为棒状或球状结构的佐证.在995 ~1 133 cm-1及447 ~485 cm-1处观察到的系列峰为SO42-伸展振动峰,这些峰在PFS 与PSF1 中的峰位差别都可以归属为受临近基团的影响不同造成的,也是PSF1 中有PS 参与Fe 络合的结果.PFS 中415 cm-1处的强峰可归属为Fe—OH 的弯曲振动吸收峰[13].
PFA,987 cm-1处的弱吸收峰是Fe—O—Al键的弯曲振动特征峰,说明Fe 参与了Al 的聚合,但是由于Fe 的含量很少导致该峰强度较弱.
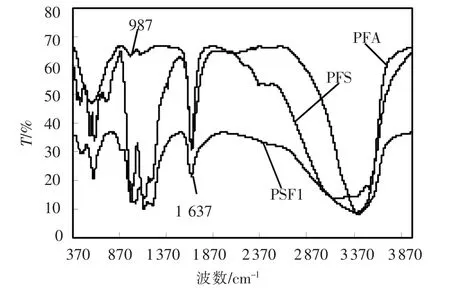
图6 PSF1、PFS 及PFA 的红外光谱图
PFS、PSF 及PFA 分别是单纯高分子、阳离子与阴离子复合高分子及不同金属复合高分子铁类混凝剂,基本代表了铁类混凝剂的研究及应用前沿,由于传统混凝剂性能较差,这里不做讨论.
混凝剂混凝性能的优劣取决于其微观性质,而PSF 独特的微观特征正是其具有优异混凝性能的根本原因.结合混凝剂微观品质及混凝效果,可以认为低温低浊水的混凝机理是以电中和/脱稳为前提条件,以架桥为必要条件,因为PFA 的Zeta电位平均值最高,几乎是PSF1 的21 倍,但是PSF1的混凝效果却远远优于PFA,这完全归功于PSF1非常大的形态尺寸以及枝杈形态的结构,因为PSF1 平均粒径是PFA 的11 倍多.而PFS 的Zeta电位和平均粒径都比较低,所以效果最差.
3 结 论
1)对PS 制备过程进行数学建模是工业制备优质PSF 的基础条件.采取多项式分段建模时,t-pH 曲线拟合效果最好,而t-温度的模型精度相对较差.较大误差都出现在曲线拐点处,这是多项式建模的缺陷.
2)将具有一定聚合度的PS 的pH 调低后,部分PS 发生解聚,但解聚后的状态完全不同于初始同样低pH 值的形态,这可能是采用酸化剂来延缓PS 聚合过程的本质原因.
3)混凝剂混凝性能的优劣取决于其微观性质,PSF 独特的微观特征正是其具有优异性能的根本原因.结合微观品质及混凝效果,可以认为低温低浊水的混凝机理是以电中和/脱稳为前提条件,以架桥为必要条件.
[1]HARRINGTON C R,WISCHIK C W,MCARRHUR F K,et al.Alzheimer’s-disease-like changes in tau protein processing:Association with aluminum accumulation in brains of renal dialysis patients[J].The Lancet,1984,343:993-997.
[2]HASEGAWA T,HASHIMOTO T,ONITSUKA T,et al.Characteristics of metal-polysilicate coagulants[J].Water Sci Technol,1991,23(7/9):1713-1223.
[3]HASHIMOTO K,HASEGAWA T,ONITSUKA T,et al.Inorganic polymer coagulants of metal-polysilicate complex[J].Water Supply,1991,9:65-70.
[4]OHNO K,UCHIYAMA M,KAMEI T,et al.Practical design of flocculator for new polymeric inorganic coagulant-PSI[J].Water Sci Technol,2004,4(1):67-75.
[5]BAYLIS J R.Silicates as aid to coagulant[J].J AWWA,1937,9:1355-1396.
[6]王东田.聚硅酸铝混凝剂的研究与应用[D].哈尔滨:哈尔滨建筑大学,1998.
[7]戴安邦,陈荣三,季明德,等.硅酸聚合理论的研究[J].南京大学学报,1964,8(1):81-86.
[8]许韵华,杨玉国,王永生,等.酸性介质对硅酸聚合胶凝的影响[J].武汉大学学报,2005,51(2):177-180.
[9]卢涌泉,邓振华.实用红外光谱分析[M].北京:电子工业出版社,1989.
[10]林树昌,曾泳淮.分析化学(仪器分析部分)[M].北京:化学工业出版社,1994.
[11]于惠,高宝玉,岳钦艳,等.红外光谱法研究聚硅氯化铝混凝剂的结构特征[J].山东大学学报,1999,34(2):198-201.
[12]康思琦,马晓鸥,刘小军,等.新型混凝剂含硼聚硅酸硫酸铁的结构分析[J].精细化工,2000,17(8):459-462.
[13]中香西尔,索罗曼P H.红外光谱分析100 例[M].北京:科学出版社,1984.
猜你喜欢
中国造纸(2022年3期)2022-07-21
昆钢科技(2021年4期)2021-11-06
食品与发酵工业(2021年14期)2021-08-02
工程建设与设计(2021年10期)2021-04-01
山西建筑(2020年2期)2020-01-09
材料科学与工程学报(2016年2期)2017-01-15
中国塑料(2016年10期)2016-06-27
河北工业大学学报(2016年6期)2016-04-16
造纸化学品(2015年4期)2015-11-04
天然产物研究与开发(2014年1期)2014-04-27
