
FC(O)O2的结构及其自由基与NO反应的微观机理研究*
2010-09-08 06:05周翔张萱刘爱芬曾祥华
物理学报 2010年7期
周翔 张萱 刘爱芬 曾祥华
(扬州大学物理科学与技术学院,扬州225002)
(2009年9月20日收到;2009年10月21日收到修改稿)
FC(O)O2的结构及其自由基与NO反应的微观机理研究*
周翔 张萱 刘爱芬 曾祥华†
(扬州大学物理科学与技术学院,扬州225002)
(2009年9月20日收到;2009年10月21日收到修改稿)
用密度泛函理论(DFT)和哈特里-福克(HF)从头计算方法和半经验势方法等研究了FC(O)O2的结构和振动性质.在DFT中采用B3LYP方法,在6-311G(d)基组上对FC(O)O2自由基与NO反应的微观过程进行了分析.首先给出了各反应物、中间体、过渡态和生成物的几何构型,然后计算了它们的能量和频率,通过频谱分析得到反应的中间体和过渡态信息,即FC(O)O2与NO反应为多反应通道,势垒高度和反应速度给出主要通道是FC(O)O2+ NO→bM→bT→FC(O)O+NO2,主要产物是自由基FC(O)O和NO2.
密度泛函理论,FC(O)O2,NO,反应机理
PACC:8220H
1. 引言
自南极洲臭氧空洞产生以来,臭氧层的损耗就成为全球普遍关心的环境问题,缺少臭氧层会引起严重的紫外辐射灾害.科学家们研究发现ClO,NOx,HOx等自由基对臭氧损耗影响很大.另一方面,随着碳氟氯烃类物质(CFCs和HFCs)在工业中的广泛应用,这类物质在大气层中会降解产生氯原子[1]或氟代甲羧酸自由基FC(O)O[2],这些自由基的存在在臭氧的耗损反应中起到催化的作用[3].1994年Dibble和Francisco等[4—8]提出在降解过程中FC(O)O2与NO反应是生成FC(O)O和NO2的最终反应,1996年Francisco等[9]详细给出降解过程为CF2O+hν→FCO+O2→FC(O)O2+NO→FC(O)O.两篇文献都提到FC(O)O2与NO是产生自由基FC(O)O的重要反应,但是都没有进行深入研究.在实验上要真实模拟大气环境的过程比较困难,在前面的工作中[10],我们对FC(O)O与NO的反应机理进行了研究,得到其与实验观测的现象一致的研究产物,即其主要产物是自由基FNO和CO2.为研究氮循环对臭氧的影响,本文利用量子化学方法,研究FC(O)O2自由基与NO反应的微观机理,这为进一步了解碳氟氯烃类物质降解过程及其产物对环境的影响都有重要意义.
2. 计算方法
目前实验中普遍采用矩阵谱仪、红外谱仪和气相谱仪三种方法,理论计算方面主要包括较为精确的量子化学计算、采用密度泛函理论(DFT)和哈特里-福克(HF)从头计算方法和半经验势方法等. DFT主要核心是利用电子密度取代波函数作为研究的基本量,根据Fermi统计,Thomas和Fermi推导出均匀多电子系统的基态能量可以表示为电子密度分布的泛函形式,这是密度泛函理论的基础,相关的理论[11,12]使DFT得到广泛应用.本文首先使用分子图形方法编辑所设想的模型物,对设计出来的所有构型首先在HF/STO-3G水平上进行结构预优化,再在HF/6-31G*水平上进行优化,然后选用不同方法、不同基组的配对(HF和B3LYP方法,6-31G*,6-311G*基组),对设计好了的结构进行优化和频率计算.最后在得到的优化结构基础上计算该构型的振动频率、振动模式、红外谱线相对强度.全部计算选用量子化学Gaussian03程序完成.在DFT中采用B3LYP方法,对FC(O)O2自由基与NO反应的反应过程进行研究.首先在6-311G(d)基组上全参数优化了FC(O)O2自由基与NO反应中各反应物、中间体、过渡态和产物的几何构型,随后通过对振动分析及内禀反应坐标(IRC)计算给出真实的中间体和过渡态.同时计算了各种结构的能量、反应过程中各种可能通道的活化能和活化熵.最后还根据过渡态理论获得了各反应通道的速度常数.除反应速率常数外,其余计算都在Gaussian03软件平台上完成.
3. 结果讨论
3.1. FC(O)O2的振动分析
首先使用分子图形方法编辑所设想的模型物,对设计出来的所有构型首先在HF/STO-3G水平上进行结构预优化,再在HF/6-31G*水平上进行优化,然后选用不同方法、不同基组的配对(HF和B3LYP方法,6-31G*,6-311G*基组),对设计好了的结构进行优化和频率计算.最后在得到的优化结构基础上计算该构型的振动频率、振动模式、红外谱线相对强度.优化得到的FC(O)O2结构如图1所示,其能量为-363.57817243 kJ/mol,属于Cs群,也是平面结构.其键长键角见表1.FC(O)O2共有5个原子,自由度为9,所以它有9种振动模式,表2给出了FC(O)O2的所有振动模式的频率,每一个振动模式的大致归属,以及红外强度和拉曼活性.从表2可以看出,FC(O)O2振动模式可以分为三类: 1)边缘原子的出平面振动、角振动.a模式属于边缘原子的出平面振动,b,c,d模式属于角振动,对应低频段.2)中心C原子的出平面振动.e模式属于中心C原子的出平面振动,对应中频段.3)键的伸缩振动.f,g,h,i模式都属于键的伸缩振动,对应高频段.图2,图3分别给出了FC(O)O2的频率在0—2500cm-1的红外光谱和拉曼光谱,图4给出了FC(O)O2的振动图.由图可见最强的振动对应于伸缩振动i.
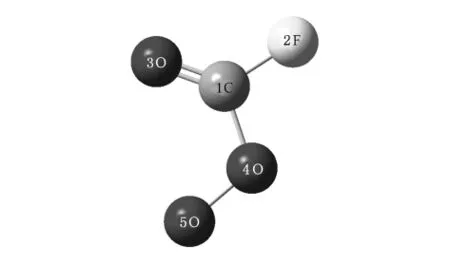
图1 FC(O)O2的优化结构

表1 FC(O)O2的键长、键角
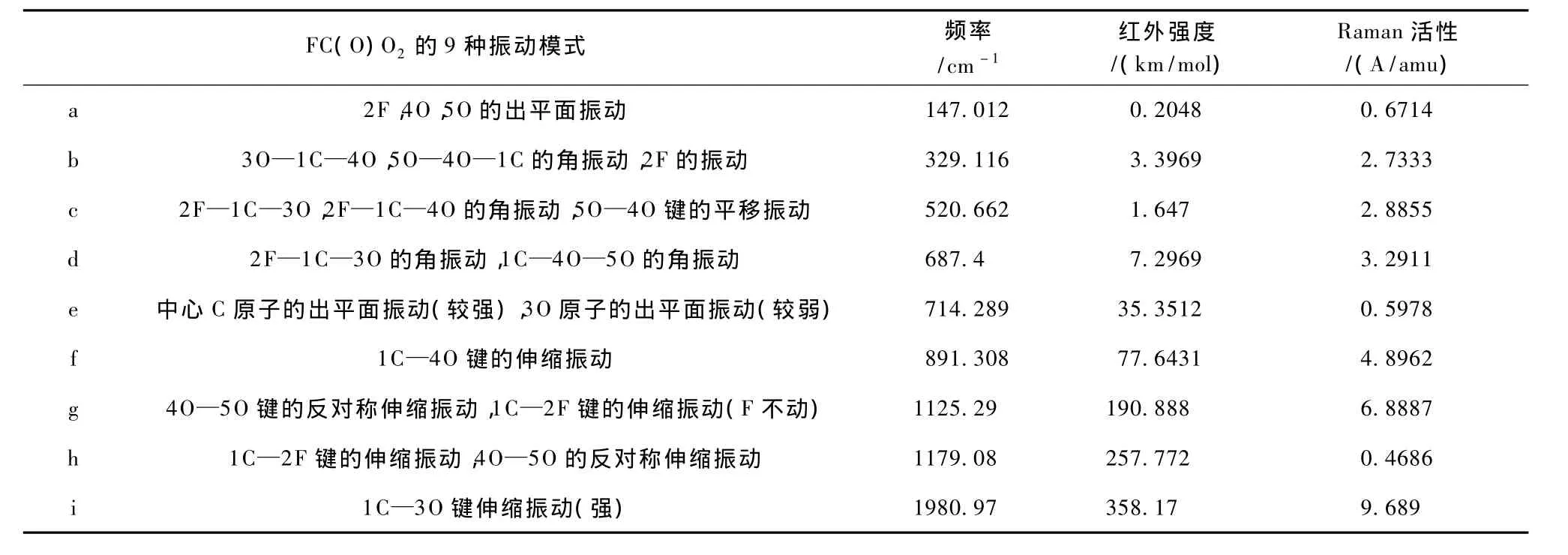
表2 FC(O)O2的振动分析表
3.2. FC(O)O2与NO反应
对FC(O)O2自由基与NO反应进行计算,分析各反应通道的微观机理.通过计算,优化得到的中间体、过渡态及产物的分子构型及参数如图5所示.

图2 FC(O)O2的红外光谱

图3 FC(O)O2的Raman光谱

图4 FC(O)O2的振动图(箭头表示振动方向)(a)—(i)分别对应表2中的振动模式a—i
化学反应具有随机性,对于FC(O)O2与NO的反应,FC(O)O2中F,O1和O3原子处于分子边缘部分,都有可能与NO的氮原子和氧原子成键.然而通过计算,没有发现NO攻击FC(O)O2中F和O1原子所形成的反应通道,只可能是NO与O3原子成键.最终结果表明该反应存在多种可能的反应通道,主要有如下四部分:

图5 FC(O)O2与NO反应中间体、过渡态及产物的分子构型及相应参数(键长单位:;键角单位:(°))
1)NO中的N原子去进攻FC(O)O2自由基中的O3原子,形成一个稳定的中间体aM,接着N原子与O2原子争夺O3原子,O3原子更倾向与N原子靠近距离逐渐变短而形成稳定的N—O3键,O2—O3键逐渐拉长而断裂形成了过渡态aT.最终FC(O)O2自由基失去一个O而变成FC(O)O,NO则得到一个O形成NO2.
2)开始同样是N原子进攻O3原子,形成稳定的中间体bM后有两种变化趋势,一种就是N—O3键逐渐变短,O2—O3键渐渐拉长而后断裂形成过渡态bT,最后形成FC(O)O和NO2.还有一种可能如图6所示,图6(a)是中间体bM的一个N和O原子的摇摆振动模式,频率为857.9cm-1.振动过程中会出现图6(b)的情况,O2—O3键在振动过程中键长是增大的,这时N原子不仅会与O4和O3成键,同时也有可能与O2原子成键,从而形成NO3结构.这样反应生成物就是FCO和NO3.
3)NO中的O原子攻击FC(O)O2自由基中的O3原子,形成中间体cM.随后O3原子会离开FC(O)O2所在的平面向上翻转,改变原来FC(O)O2自由基的平面结构.O4—O3键伸长断裂形成过渡态cT.最终生成FC(O)ONO2.
4)也是NO中的O原子攻击FC(O)O2自由基中的O3原子,形成另一个中间体dM.图6(c)是中间体dM一个振动模式,频率为610.9cm-1.不停振动过程中会出现图6(d)的状态,N原子可能和三个O原子结合,O1原子和C,F原子结合生成FCO,最终产物是FCO和NO3.当然dM变化也可能出现类似cM的状况,O3原子会离开FC(O)O2所在的平面向上翻转,O4—O3键伸长断裂形成过渡态dT.最后的生成物也是FC(O)ONO2.在所有中间体、过渡态和产物中,除了FC(O)ONO2具有Cs对称性,其余属于C1群.

图6 中间体bM和dM的振动图像(a),(b)为中间体bM的摇摆振动;(c),(d)为中间体dM的振动
通过对FC(O)O2自由基与NO反应的产物、中间体的结构振动分析,发现力常数矩阵本征值全为正,说明有势能面上的稳定点;对过渡态aT,bT,cT和dT的振动分析发现力常数矩阵本征值均唯一的负值,对应的虚频率分别为-711.1,-168.5,-206.8和-52.4cm-1.根据化学反应过渡态判据理论[13,14],这些态是真实过渡态.
3.3. 对FC(O)O2自由基与NO反应路径的能量分析
为说明FC(O)O2与NO的反应过程,表3列出了相应的反应物、中间体、过渡态和产物的能量、第一个振动频率,并给出了在B3LYP方法,6-311G(d)基组下FC(O)O2与NO为参考的相对能量ΔE.
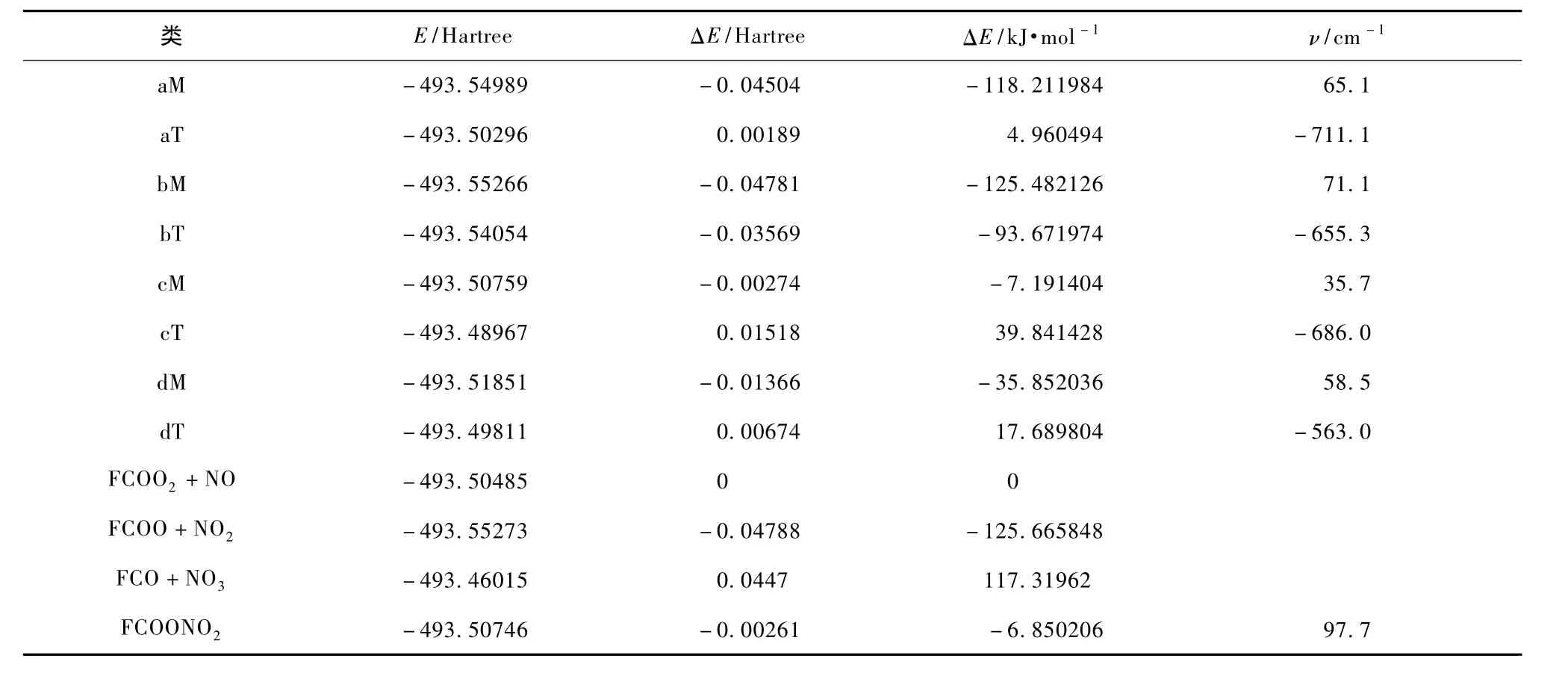
表3 反应各个驻点的相对能量
根据相对能量ΔE,分别计算出如下4类反应通道的反应活化能.
第1类FC(O)O2+NO→aM→aT→FC(O)O +NO2反应通道,aM经过过渡态aT反应活化能为123.2 kJ/mol,由于是单步反应,这个值也是控制步骤反应活化能;
第2类FC(O)O2+NO→bM→bT→FC(O)O +NO2反应通道,bM经过过渡态bT反应活化能为31.8 kJ/mol,而bM和dM可能直接生成FCO+ NO3,则没有经过过渡态;
第3类FC(O)O2+NO→cM→cT→FC(O) ONO2反应通道,cM经过过渡态cT合成反应活化能为46.9 kJ/mol;
第4类FC(O)O2+NO→dM→dT→FC(O) ONO2反应通道,反应通道控制步骤反应活化能为53.5 kJ/mol.根据反应路径随反应进程的变化情况,绘出了相应的能级示意图,如图7所示.

图7 FC(O)O2+NO反应中的能量关系
由表3和图7可知FC(O)O2和NO的几条反应通道,只有中间态bM和dM直接生成FCO+NO3的反应通道是吸热反应,其余的几个通道都是放热反应,所以这两个通道不是优势通道.而且生成物FCO是降解反应链中的重要物质,在大气中氧气浓度很高的环境下易重新产生FC(O)O2,这都表明这两个反应通道概率不大.第3和第4类生成FC(O) ONO2反应通道控制步骤活化能不大,而且均为放热反应,分别是46.9和53.5 kJ/mol,但是产物能量也比较高,所以不是两个理想的反应通道,而且FC(O)ONO2的逆反应(分解为FC(O)O2+NO)也相当快[15],所以这两个通道并非最佳通道.第1类和第2类生成FC(O)O+NO2反应通道,控制步骤活化能分别为123.2和31.8 kJ/mol,生成物FC(O) O+NO2能量相对最低,应该是两个优势反应通道.而FC(O)O2+NO→bM→bT→FC(O)O+NO2反应通道控制步骤活化能只有第1类反应的1/3.因此从能量角度分析这个反应通道概率最大,是最主要反应通道,这也和相关文献一致[4—9].
3.4. 反应速率常数计算
根据经典的过渡态理论,利用优化所得的活化能和活化熵的数据,用公式k=来计算各反应通道的速率常数.式中kB是玻尔兹曼常数,是约化普朗克常数,T是绝对温度,R是气体常数,ΔS是活化熵,ΔE是活化能.不经过过渡态的反应通道为自由基的无垒反应过程,反应速率极快,这里就不考虑,经过过渡态的反应通道都是异构化反应,为一级反应.本次计算是在298.15 K,一个标准大气压下进行.活化能为中间体与过渡态之间的能量差值,计算结果见表4.

表4 过渡态理论计算得到的反应速率常数
由表4可见,bM转化为bT的步骤进行得非常快,反应速率常数为2.95×107s-1.这比其他通道的反应都快了1000倍以上,而且该通道产物能量很低,因此bM→bT的通道是最优反应通道.cM→cT和dM→dT两个通道的速度都比较快,但它们的逆反应速度也非常快[15],FCOONO2的生成和分解能够很快达到动态平衡.aM异构化为aT的反应速率常数为3.08×10-11s-1,虽然比bM→bT的反应慢了很多,但是也并不是没有反应的可能,这个通道也应该在整个过程中存在.因此aM→aT和bM→bT一样也是一个比较优越的反应路径,但最终两个通道的产物都为FCOO+NO2,这又一次证实了该反应过程的真实性.
4. 结论
通过对FC(O)O2和NO反应机理的理论研究发现:该反应4个主要反应通道分别为

通过对反应通道的活化能和反应速率常数的计算发现:FC(O)O2自由基和NO反应的主要反应通道为FC(O)O2+NO→bM→bT→FC(O)O+NO2,整个过程是一个放热反应,主要反应产物为FC(O)O和NO2.其他反应通道产物有FC(O)ONO2,NO3和FCO.
随全球极端气候的快速变化,人类对生存的环境越来越担忧,因为温室效应导致的后果已影响到人类的生存.2009年11月有100多个国家参加的联合国气候变化大会达成了不具法律约束力的《哥本哈根协议》,其目的在于控制温室效应,减少碳排放.而臭氧层的损耗是导致气候变暖的主要原因.本文着重讨论的过氧氟代甲羧酸自由基与NO反应生成的氟代甲羧酸自由基FC(O)O,它将与NO反应生成FNO,而FNO在太阳的紫外线辐射作用下会分解为自由氟原子,它对臭氧损耗起着重要的作用.我们只对温室气体中的氮类氧化物、含氟类物质的作用过程进行了初步研究.大气层中臭氧的损耗比较复杂,在太阳紫外辐射作用下各种温室气体之间的反应会加剧,产生的物质对臭氧损耗将更加严重,这需要更多、更深入的研究,如建立相应的模拟反应实验室等对此进行系统的研究.
[1]Cicerone J R 1987Science 237 35
[2]Maricq M M,Szente J J,Li Z,Francisco J S 1993 J.Chem. Phys.98 784
[3]Francisco J S,Goldstein A N,Li Z,Zhao Y,Williams I H 1990 J.Phys.Chem.94 4791
[4]Dibble T S,Francisco J S 1994 J.Phys.Chem.98 10374
[5]Maricq M M,Szente J J,Dibble T S,Francisco J S 1994 J. Phys.Chem.98 12294
[6]Dibble T S,Francisco J S 1994 J.Phys.Chem.98 1694
[7]Dibble T S,Francisco J S 1994 J.Phys.Chem.99 5010
[8]Dibble T S,Francisco J S,Deeth R J,Hand M R,Williams I H 1994 J.Chem.Phys.100 459
[9]Joseph S,Francisco J S 1996 Acc.Chem.Res.29 391
[10]Zhao J,Zeng X H,Cui L,Xu X L 2008 Acta Phys.Sin.57 7349(in Chinese)[赵江、曾祥华、崔磊、徐秀莲2008物理学报57 7349]
[11]Hohenberg P,Kohn W 1964 Phys.Rev.B 136 864
[12]Kohn W,Sham L J 1965 Phys.Rev.A 140 1133
[13]Wei Y Y,Li J 2004 The Conspectus of Chemical Reaction Mechanism(Beijing:Science Press)(in Chinese)[(魏运洋、李建2004化学反应机理导论(北京:科学出版社)]
[14]Zhao X S 2004 The Conspectus of Chemical Reaction Theory (Beijing:Peking University Press)(in Chinese)[赵新生2003化学反应理论导论(北京:北京大学出版社)]
[15]Zhang J S,Meng Q X,Li M 2005 Acta Chimica Sinica 63 686 (in Chinese)[张金生、孟庆喜、李明2005化学学报63 686]
PACC:8220H
*Project supported by the Science and Technology Program of Jiangsu Province,China(Grant No.BG2007026).
†Corresponding author.E-mail:xhzeng@yzu.edu.cn
Structure of FC(O)O2and the mechanism of its reaction with NO*
Zhou Xiang Zhang Xuan Liu Ai-Fen Zeng Xiang-Hua†
(College of Physics Science and Technology,Yangzhou University,Yangzhou225002,China)
(Received 20 September 2009;revised manuscript received 21 October 2009)
Density functional theory was performed to study the reaction mechanism of the reaction of FC(O)O2with NO.The geometric configurations of reactants,intermediates,transition states and products were optimized by B3LYP method at 6-311G(d)level.The energies of stationary points along the pathway were also calculated.Intermediates and transition states were confirmed by the results of vibrational analysis.From the results of the mechanism of the reaction of FC(O)O2with NO,we found that the reaction of FC(O)O2+NO has four pathways and several steps.Comparing the four pathways' activation energies,we can find that the pathway FC(O)O2+NO→bM→bT→FC(O)O+NO2is the main reaction pathway and the main products are FC(O)O radical and NO2,which is in good agreement with the result reported in the literature.
density functional theory,FC(O)O2,NO,reaction mechanism
book=315,ebook=315
*江苏省科技计划(批准号:BG2007026)资助的课题.
†通讯联系人.E-mail:xhzeng@yzu.edu.cn
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
数学年刊A辑(中文版)(2021年1期)2021-06-09
汕头大学学报(自然科学版)(2020年4期)2020-12-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
新高考·高一物理(2016年3期)2016-05-18
股市动态分析(2015年12期)2015-09-10
无机化学学报(2014年12期)2014-02-28
