
毛细管电泳法测量清热化毒丸中盐酸小檗碱的含量
2010-08-28 03:32王维庭房娟娟闫滨
中国现代药物应用 2010年13期
王维庭 房娟娟 闫滨
清热化毒丸由连翘、青黛、黄连、大黄、菊花等15味药物组成,具有清火化毒,消肿止痛的作用。用于小儿身热烦躁,咽喉肿痛,口舌生疮,皮肤疮疖口臭便秘,疹后余毒未尽等症状。盐酸小糪碱是清热化毒丸中主要活性成分之一,以抗菌消炎为主。
近年来分析小檗碱型生物碱的方法分别有反向高效液相色谱法[1],离子交换色谱法[2],胶束色谱法[3],薄层色谱-荧光分光光度法等。但此样品成分复杂,且盐酸小檗碱为季胺类生物碱,碱性强,极性大,往往被色谱柱吸附,影响峰形,对色谱柱造成很大污染,高效毛细管电泳(HPCE)以其高效灵敏、分离速度快、样品量小、消耗仅微量等优点已越来越多的用于中药成分分析及测定中。本文通过对清热化毒丸中盐酸小糪碱的毛细管电泳条件进行摸索,建立了盐酸小糪碱含量测定的高效毛细管电泳方法。
1 仪器与试剂
1.1 仪器 Beckman P/ACE 5500型高效毛细管电泳仪(美国Beckman公司),配备UV检测器,Chemical Station工作站;酸度计Model PHS-2CA(上海大中分析仪器厂);超声清洗器KQ-500E(江苏昆山市超声仪器有限公司);万分之一电子天平(METTLER-TOLEDO中国);涂层熔融石英毛细管柱(47 cm×75 m,河北永年光导纤维厂)。
1.2 试剂 盐酸小檗碱对照品(批号:110713-200609)购于中国药品生物制品检定所;甲醇为色谱纯(天津市广成化学试剂有限公司),水为自制重蒸馏水,其他试剂均为分析纯。所需清热化毒丸(北京同仁堂股份有限公司同仁堂制药厂,批号:9013395),购自药店。
2 含量测定
2.1 溶液制备
2.1.1 对照品溶液的制备 精密称取盐酸小檗碱对照品适量,加甲醇制成1 mg/ml的对照品溶液,4℃保存备用。
2.1.2 供试品溶液的制备 取清热化毒丸2丸,剪碎,称取3.0065 g置于100 ml具塞锥形瓶中,精密加入甲醇50 ml,称重后超声(功率250 W,频率30 kHz)处理30 min,放冷,称定重量,必要时用甲醇补足失重,过滤。滤液自然挥干后用甲醇溶解,转移至5 ml容量瓶中,加入2 ml缓冲液后用甲醇定容,最后用0.45 μm有机膜过滤备用。
2.2 电泳条件 涂层熔融石英毛细管柱(47 cm×75 m,有效长度为40 cm);检测波长为200nm,进样:氮气压力(586 KPa)自动进样5 s,分离电压为30KV,柱温25℃,缓冲液体系为60 mmol/L磷酸缓冲溶液-甲醇(65:35),调节 pH值为3.2。
2.3 线性关系考察 精密称取盐酸小檗碱对照品适量分别于10 ml容量瓶中,加入2 ml缓冲溶液,用甲醇定容至刻度得 0.05,006,0.07,008,0.09,0.10 mg/ml的对照品溶液,依次进样,以对照品浓度为横坐标对峰面积做回归分析,得回归方程为Y=1000000x-8143.9,r=0.9993。结果表明盐酸小檗碱在0.05~0.10 mg/ml范围内线性关系良好,(对照品色谱图见图1)。

图1 盐酸小糪碱毛细管电泳色谱图
2.4 精密度试验 取0.1 mg/ml盐酸小糪碱对照品溶液,在上述电泳条件下,连续进样5次,以峰面积计算RSD为0.50%,表明在此条件下精密度良好。
2.5 稳定性试验 取供试品溶液一份,分别于0,2,4,8,10 h测定样品的峰面积,求得RSD值为1.29%,实验表明供试品在10 h内稳定性良好。
2.6 回收率测定 取5份已知含量的供试品溶液,加入适量对照品,按“2.1.2”项下的方法制备,通过HPCE进行测定,结果见表1。

表1 清热化毒丸中盐酸小糪碱加样回收率(g,mg,%)
2.7 样品含量测定 按“2.1.2”项方法制备供试品溶液3份,在上述电泳条件下,每份进样两次取平均值,得盐酸小檗碱的含量测定结果为0.1357 mg/g(样品色谱图见图2)。
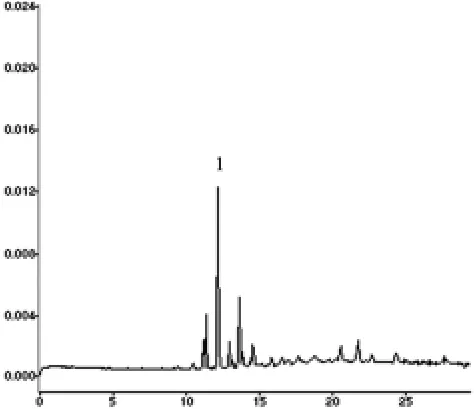
图2 清热化毒丸中盐酸小糪碱毛细管电泳色谱图
3 讨论
3.1 缓冲溶液的选择 由于盐酸小檗碱为季胺类生物碱,带有正电荷,用磷酸氢二钠做为缓冲液可使其保持带有正电荷,不同的缓冲液及不同的比例会影响成分之间的分离度。本实验先后在硼砂-甲醇及磷酸二氢钠-甲醇不同浓度缓冲体系下进行电泳分离,发现60 mmol/L磷酸二氢钠-甲醇(65:35)的缓冲体系分离度较高,可达到良好的分离效果。
3.2 pH值的选择 在毛细管电泳中,电渗流对pH值有较强的依赖性,直接影响待测成分的出峰时间,pH值降低,缓冲液呈酸性时,电渗流减弱,成分出峰时间延后,pH值升高,缓冲液偏碱性时,电渗流增强,成分出峰时间提前。并且离子的电泳淌度跟有效电荷成正比,极易受缓冲溶液pH值影响,因此缓冲溶液的pH值调节与控制是优化分离的重要对策。同时缓冲溶液的pH值也会影响离子的有效电荷,从而间接影响分离效果。在硼砂-甲醇(85:15)缓冲体系(pH=9.0)中,样品的出峰时间短,但未能达到基线分离。本实验又分别考察了磷酸二氢钠缓冲体系(pH值分别为3.0、4.0、5.0)对分离效果的影响。结果表明,pH值为3.0时有良好的分离效果。
3.3 分离电压的选择 本试验考察了电压为15 KV~30 KV时的影响。结果表明:电压低时,基线稳定,但样品的分析时间延长,峰形变宽;随着电压的增加,样品的迁移时间缩短,峰型窄,分离效果好,但基线噪音有所增大。基于对迁移时间和基线的综合考虑,本试验最终选用30 KV作为分离电压。
3.4 毛细管有效柱长的选择 熔融石英毛细管柱长是改变出峰时间的主要因素之一,一般目标成分的出峰时间控制在15 min以内为宜。本实验考察了有效长度为40 cm、50 cm、60 cm的毛细管对出峰时间的影响,结果表明,三者对于目标组分均有良好的色谱峰形和分离度,但40 cm柱长出峰时间在5 min左右,较为快速,因此优先选择40 cm柱长毛细管。
综上所述,在目前分析黄连药材及其复方制剂的较常用的含量测定方法中,高效液相色谱法应用最为广泛,薄层层析方法已经较为少见。高效液相色谱法分离具有重现性好,分离效果稳定、灵敏度高的优点,但是由于小糪碱是季胺碱,碱性强,极性强,易离子化,且易使液相色谱柱中的ODS染色,需要大量溶剂冲洗且耗时较多,而且由于黄连中含多种季胺碱,极性类似,液相色谱不易将其分开,相互之间易造成干扰。HPCE的区带电泳模式,由于使用熔融石英毛细管空管,不怕污染和堵塞,易清洗,且有分离效率高,样品需要量极少的特点,仅使用简单少量的缓冲液,几乎不产生废液,分析耗费低,样品预处理简单,几乎可称为绿色分析方法,尤其适合于基质比较复杂,结构近似的药用植物成分的分析。在本实验中,可同时分离黄连中的7个季胺生物碱,专属性和分离度良好,可用于该类成分的指纹图谱研究。HPCE既可结合质谱技术建立复方药物指纹图谱,又可进行多种有效成分或指标成分定量,还可用于体内药物分析,开展有效成分研究。HPCE分离分析药用植物成分的发展具有极为广阔的前景。
[1]国家药典委员会.中华人民共和国药典(一部).北京:化学工版社,2005:705.
[2]文欣欣,刘丹华,余陈欢,等.薄层色谱-荧光分光光度法测定黄连及香连丸中盐酸小檗碱的含量.中国中医药信息杂志,2008,15(10):54.
[3]余为,胡薏冰.高效毛细管电泳法测定紫黄止血消炎粉中盐酸小檗碱的含量.中国实用医药,2008,3(15):73.
猜你喜欢
食品工业科技(2021年23期)2021-12-16
现代临床医学(2021年4期)2021-07-31
西南农业学报(2020年8期)2020-12-10
食品安全导刊·中旬刊(2020年6期)2020-07-28
食品安全导刊(2020年17期)2020-07-18
中成药(2018年12期)2018-12-29
中成药(2018年3期)2018-05-07
科学中国人(2016年30期)2016-07-14
中国粮油学报(2016年1期)2016-02-06
杂草学报(2015年2期)2016-01-04
