
柴胡炮制后皂苷成分的变化分析
2010-05-26 06:14:40孙秋实
中成药 2010年5期
陈 帅, 李 燕, 孙秋实, 杨 方, 李 悦, 吴 彤
(上海医药工业研究院,上海 200040)
柴胡为伞形科植物柴胡Bupleurum chinenseDC.或狭叶柴胡Bupleurum scorzonerifoliumWild.的干燥根,味苦,微寒,归肝、胆经,具有疏散退热,舒肝、升阳的作用[1]。历代本草和现代中医的肝病用药中多以柴胡为君药。但柴胡的不同炮制品治疗病症侧重点不一,在用于治疗肝病时,常用其醋制品,2005版《中国药典》中就收载了醋炙柴胡这一炮制品种。对于中药炮制,国内研究主要集中在炮制原理的探究和炮制方法的改进等方面[2],对炮制前后的药材化学成分变化的研究较少,仅局限于针对某一种成分的变化进行研究,如总皂苷、挥发油[3]等。本实验对柴胡炮制后及皂化反应后皂苷成分的变化进行了系统的分析研究,结果显示炮制后,柴胡中总皂苷的含量都有所下降,柴胡皂苷b2的含量都大幅增加,柴胡皂苷a、c、d以及a+c+d的含量略微有所减少。皂化反应后,柴胡皂苷a+c+d的含量变化很小,柴胡皂苷b2的含量都大幅增加。揭示了柴胡炮制后皂苷类化合物变化规律,为柴胡饮片的炮制及质控提供较为全面的参考。
1 仪器、材料和试剂
1.1 仪器、材料 UV-8500紫外分光光度仪;HP1100高效液相色谱系统:G1322A脱气机,G1316A柱温箱,G1311A四元泵,G1314A VWD紫外检测器。
1.2 试剂 甲醇、乙腈、乙醇、氢氧化钠、氢氧化钾、磷酸、碳酸钠、对二甲氨基苯甲醛、氨水、正丁醇、磷酸二氢钾均为分析纯;水(超纯水)。柴胡皂苷a、c、d、b2均为本实验室自制(纯度大于98%)。
1.3 药材 见表1。

表1 柴胡及醋制柴胡药材编号及产地Tab.1 Serial numbers and places of production of the medicinal materials and the processed bupleurum
2 方法与结果
2.1 柴胡皂苷b2的含量测定
2.1.1 分析条件
色谱柱:Diamonsil C18(250 mm×4.6 mm);流速:1 mL/min;柱温:35℃;检测波长:248 nm。见表2。

表2 乙腈-水梯度洗脱条件Tab.2 Gradient elution program of acetonitrile and water
2.1.2 提取方法的考察
2.1.2.1 提取方式的选择
取CH-5号药材0.1 g两份,精密称量,置于茄形瓶中,一份精密的加入20 mL 5%氨水甲醇溶液于80℃水浴回流2 h,过滤,蒸干,精密加入5 mL甲醇溶液定容;另一份于试管中超声两次,每次加入5%氨水甲醇溶液10 mL,超声1 h,离心,将两次上清液合并蒸干,精密加入5 mL甲醇溶液定容,按2.1.1项色谱条件进行分析,实验结果显示柴胡皂苷b2回流的提取效果优于超声,所以选择回流这种提取方式。
2.1.2.2 提取时间的选择
取CH-5号药材0.1 g,精密称量,置茄形瓶中,分别精密的加入20 mL 5%氨水甲醇溶液于80℃水浴回流,分别1 h、2 h、3 h、4 h,回流后过滤,蒸干,分别精密加入5 mL甲醇溶液定容,进样,按2.1.1项色谱条件进行HPLC分析。1 h、2 h、3 h、4 h提取后含量几乎没有明显的变化,为了既节约时间又使其充分的回流,并结合文献的报道,选择2 h为回流提取的时间。
2.1.2.3 提取次数的选择
取CH-5号药材0.1 g两份,精密称量,置于茄形瓶中,分别精密的加入5%氨水甲醇溶液20 mL于80℃水浴回流,一份回流2 h,过滤,蒸干,精密加入5 mL甲醇溶液,定容;另一份先回流2 h后,将上清液过滤,再加入20 mL 5%氨水甲醇溶液于滤渣中,再回流2 h,合并两次滤液,蒸干,精密加入5 mL甲醇溶液,定容,按2.1.1项色谱条件进行HPLC分析。结果显示回流一次和两次对于柴胡皂苷b2的提取效果并无明显差异,所以选择回流1次。
2.1.2.4 提取溶剂的选择
柴胡提取溶剂的选择取CH-5号药材0.1 g两份,精密称量,置于茄形瓶中,一份加入5%氨水甲醇溶液20 mL,另一份加入5%氨水95%乙醇溶液20 mL,于80℃水浴回流2 h,过滤,蒸干,精密加入5 mL甲醇溶液,定容,按2.1.1项色谱条件进行分析。结果显示对于柴胡皂苷b2的提取,加入5%氨水甲醇溶液的提取效果优于加入5%氨水95%乙醇溶液,所以对于柴胡的提取选择加入5%氨水甲醇溶液。
醋制柴胡提取溶剂的选择 取CH-5-4号药材0.1 g 4份,精密称量,置于茄形瓶中,分别精密的加入20 mL甲醇、5%氨水甲醇、95%乙醇、5%氨水95%乙醇溶液于80℃水浴回流2 h,过滤,蒸干,精密加入5 mL甲醇溶液定容,按2.1.1项色谱条件进行分析,结果显示5%氨水甲醇溶液对醋柴胡中柴胡皂苷b2的提取效果也较好。
综上,得出柴胡皂苷b2的最佳提取方法:药材粉末0.1 g,加入5%氨水甲醇溶液20 mL,于水浴回流2 h。
2.1.3 含量测定
取不同柴胡样品,按供试品溶液制备方法操作并按2.1.1项色谱条件进行HPLC分析,记录柴胡皂苷b2的峰面积,计算含量,结果见表3。见图1~4。
柴胡皂苷b2的含量测定结果显示柴胡炮制后,柴胡皂苷b2的含量从10.7%~78.2%都有不同程度的提高。
2.1.4 方法学考察
2.1.4.1 对照品溶液的制备
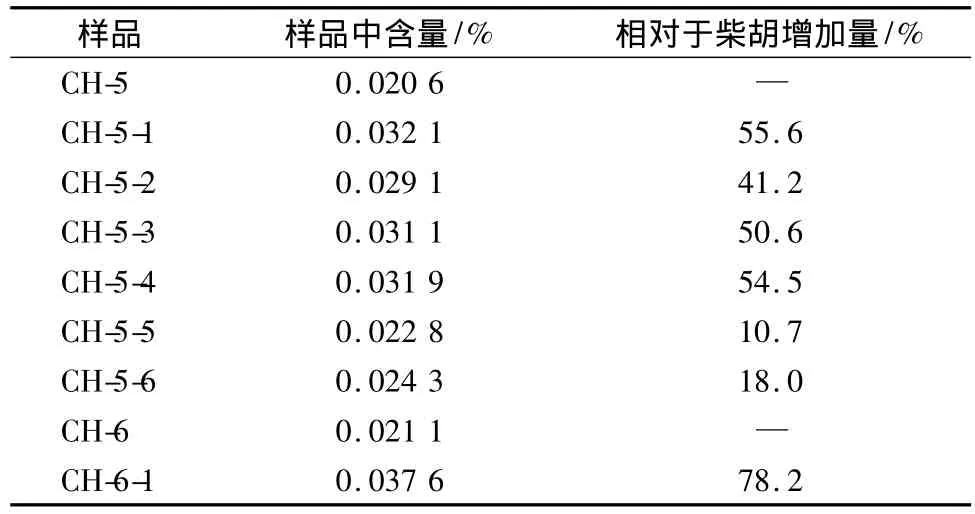
表3 不同批次柴胡中柴胡皂苷b2的含量Tab.3 Content determination of saikosaponin b2of various samples

图1 对照品柴胡皂苷b2HPLC图谱Fig.1 HPLC chromatogram of saikosaponin b2 reference substance

图2 药材CH-5中柴胡皂苷b2含量测定HPLC图谱Fig.2 HPLC chromatogram of saikosaponin b2in medicinal material CH-5

图3 药材CH-5-1中柴胡皂苷b2含量测定HPLC图谱Fig.3 HPLC chromatogram of saikosaponin b2in medicinal material CH-5-1
柴胡皂苷b2自制。取柴胡皂苷b2对照品适量,精密称量,加甲醇制成每1 mL中含0.067 8 mg的溶液,即得。
2.1.4.2 供试品溶液的制备
取柴胡药材0.1 g,精密称量,置于茄形瓶中,分别精密的加入5%氨水甲醇溶液20 mL于80℃水浴回流2 h,将滤液过滤,蒸干,精密加入5 mL甲醇溶液定容,即为供试品溶液。

图4 药材CH-5-2中柴胡皂苷b2含量测定HPLC图谱Fig.4 HPLC chromatogram of saikosaponin b2in medicinal material CH-5-2
2.1.4.3 标准曲线
取柴胡皂苷b2对照品溶液,精密吸取1.0、2.5、5.0、10.0、15.0、20.0 μL,按 2.1.1 项色谱条件进行HPLC分析,以进样量(μg)为横坐标,峰面积值(A)为纵坐标绘制标准曲线,得回归方程:A=75.6C-8.90,R2=1。结果表明柴胡皂苷b2在0.067 8~1.36 μg范围内,进样量与峰面积之间线性关系良好。
2.1.4.4 精密度
取对照品溶液,在2.1.1项色谱条件下进行测定,连续进样5次,记录柴胡皂苷b2的峰面积,并计算相对标准偏差,RSD为0.69%。
2.1.4.5 重复性
取样品CH-5-1,5份,每份0.1 g,精密称定,照供试品溶液制备方法分别制成供试品溶液,按2.1.1项色谱条件测定,记录柴胡皂苷b2的峰面积,计算相对标准偏差,RSD为0.49%。
2.1.4.6 稳定性
取样品CH-5-1,0.1 g,精密称定,照供试品溶液制备方法制成供试品溶液,按2.1.1项色谱条件分别于0、2、4、6、8 h 测定,记录柴胡皂苷 b2的峰面积,计算相对标准偏差,RSD为1.7%,表明样品在8 h内稳定。
2.1.4.7 加样回收率
取6份已知含量的同一批柴胡样品CH-5-1,各0.05 g,精密称定,分别加入对照品溶液0.067 8 mg/mL,3.7、5、6 mL,按供试品溶液制备方法制备样品试液,按2.1.1项色谱条件进样分析,记录柴胡皂苷b2的峰面积并计算其含量,平均加样回收率为97.4%,RSD为2.6%。
2.2 柴胡皂苷a、c、d的含量测定
2.2.1 分析条件
色谱柱、流动相、流速和柱温条件参考2.1.1项;检测波长:204 nm。
2.2.2 对照品溶液的制备
柴胡皂苷a、c、d均为实验室自制。分别取柴胡皂苷a、c、d对照品适量,精密称定,加甲醇制成浓度分别为 0.216、1.44、0.278 mg/mL的溶液,即得。分别取柴胡皂苷 a、c、d,0.8、0.09、1 mL 于 2 mL 量瓶中,用甲醇定容,得到混合对照品溶液Ⅰ,取混合对照品溶液Ⅰ0.5 mL于1 mL量瓶中,用甲醇定容,得到混合对照品溶液Ⅱ。即每1 mL混合对照品溶液Ⅱ中分别含柴胡皂苷 a、c、d为 0.008 64 mg、0.006 48 mg、0.013 9 mg。
2.2.3 供试品溶液的制备
取柴胡药材0.1 g于茄形瓶中,精密称定,分别加入20 mL 5%氨水-甲醇溶液于80℃水浴回流2 h,将滤液过滤蒸干,精密加入10 mL甲醇溶液定容,即为供试品溶液。
2.2.4 含量测定
取不同批次的柴胡样品,按供试品溶液制备方法操作并按2.2.1项色谱条件进行HPLC分析,记录柴胡皂苷a、c、d的峰面积,计算含量,结果见表4,图5~8。

表4 不同批次柴胡中柴胡皂苷a、c、d的含量Tab.4 Content determination of saikosaponin a、c、d of various samples

图5 对照品柴胡皂苷c、d、a HPLC图谱Fig.5 HPLC chromatogram of saikosaponin c、d、a reference substance
结果显示柴胡皂苷a+c+d的含量在炮制后含量略微有所减少,减少的程度从-0.375%~25.2%。表明原生皂苷在酸的条件下转化为次生皂苷,次生皂苷b2的含量有所增加,而原生皂苷柴胡皂苷a+c+d的含量有所减少。
2.2.5 方法学考察
2.2.5.1 标准曲线
取柴胡皂苷混合对照品溶液Ⅱ,精密吸取2.5、5.0、10.0、15.0、20.0 μL,按 2.2.1 项色谱条件进行HPLC分析,以进样量(μL)为横坐标,峰面积值(A)为纵坐标绘制标准曲线,得回归方程,结果见表5,表明柴胡皂苷 a、c、d 分别在0.216 ~1.73 μg、0.162~1.30 μg、0.350 ~2.78 μg 范围内,进样量与峰面积之间线性关系良好,符合外标法定量测定要求。
2.2.5.2 精密度
图6 药材CH-5中柴胡皂苷c、d、a含量测定HPLC图谱Fig.6 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5
图7 药材CH-5-1中柴胡皂苷c、d、a含量测定HPLC图谱Fig.7 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5-1

图8 药材CH-5-2中柴胡皂苷c、d、a含量测定HPLC图谱Fig.8 HPLC chromatogram of saikosaponin c、d、a in medicinal material CH-5-2
表5 柴胡皂苷a,c,d的线性关系测定结果
Tab.5 Results of linear ranges of saikosaponin a、c、d

指标成分 回归方程 R2 线性范围/μg+3.24 0.999 0.216~1.73柴胡皂苷c Y=20.4X+0.233 0.999 0.162~1.30柴胡皂苷d Y=57.0X柴胡皂苷a Y=46.2X+8.22 0.999 0.350~2.78
取柴胡皂苷混合对照品溶液Ⅱ,在2.2.1项色谱条件下进行HPLC分析,连续进样5次,记录柴胡皂苷a、c、d的峰面积并计算相对标准偏差,柴胡皂苷 a、c、d 的 RSD 分别为0.36%、0.63%、0.29%,表明色谱系统精密度符合要求。
2.2.5.3 重复性
取样品CH-5-2,5份,每份0.1 g,精密称量,按照供试品溶液制备方法分别制成供试品溶液,按2.2.1项色谱条件测定,记录柴胡皂苷a、c、d的峰面积,计算相对标准偏差,柴胡皂苷a、c、d的RSD分别为2.0%、1.8%、1.9%,表明方法的重复性较好。
2.2.5.4 稳定性
取样品CH-5-2,0.1 g,精密称量,照供试品溶液制备方法分别制成供试品溶液,按2.2.1项色谱条件分别于 0、2、4、6、8 h 测定,记录柴胡皂苷 a、c、d的峰面积,计算相对标准偏差,柴胡皂苷a、c、d的RSD分别为0.85%、2.1%、1.9%,表明方法的稳定性较好。
2.2.5.5 加样回收率
取5份同一批柴胡样品 CH-5-2,各0.05 g,精密称量,分别加入混合对照品溶液(Ca=0.53 mg/mL,Cc=0.098 mg/mL,Cd=0.60 mg/mL)0.65 mL、0.75 mL、0.85 mL,按供试品溶液制备方法制备样品试液,按2.2.1项色谱条件进行测定,记录柴胡皂苷a、c、d的峰面积,计算相对标准偏差,柴胡皂苷a、c、d的平均加样回收率为 94.0%、95.6%、98.9%,RSD分别为2.7%、3.2%、2.3%。
2.3 皂化后皂苷的含量测定
2.3.1 皂化方法的比较
结合文献报道的两种皂苷皂化的方法[4,5],采用以下方法进行了比较。
2.3.1.1 皂化方法Ⅰ
取柴胡药材约0.1 g,精密称量,置于茄形瓶中,加5%氢氧化钾甲醇20 mL液浸渍过夜,加热回流5 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氢钾溶液50 mL洗涤,弃水相,再以40 mL水洗涤1次,有机相蒸干,精密加入10 mL甲醇定容。用2.1.1项、2.2.1项的分析条件进行分析,得到如表6结果:
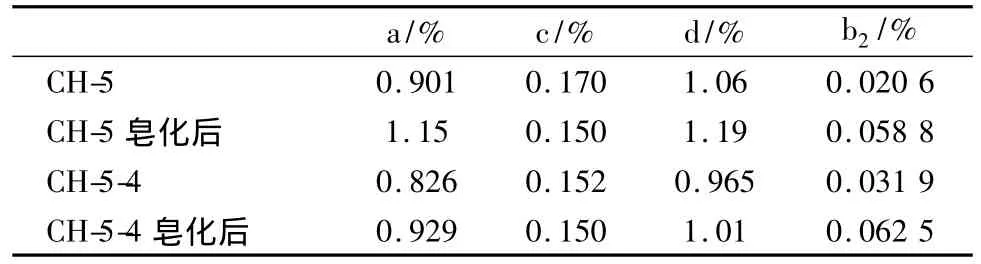
表6 皂化反应的选择Tab.6 Selection of saponated action
2.3.1.2 皂化方法Ⅱ
药材粉末1.0 g,用90%甲醇提取3次,每次15 min,溶剂量分别为 20 mL,15 mL,15 mL,合并 3 次滤液,用90%甲醇定容到50 mL,取其中的5 mL,加入氢氧化钠溶液2.5 mL于50℃水浴回流1 h,然后加入磷酸二氢钾-氢氧化钠缓冲溶液(100 mL 0.2 mol/L磷酸二氢钾和69.5 mL 0.2 mol/L氢氧化钠混合)7.5 mL,混合液过减压ODS柱,先用35%甲醇洗脱10 mL,再用甲醇洗脱,收集甲醇洗脱液10 mL。
药材粉末0.1 g,用90%甲醇提取3次,每次15 min,溶剂量分别为 2 mL,1.5 mL,1.5 mL,合并3 次滤液,用90%甲醇定容到5 mL,加入氢氧化钠溶液0.25 mL于50℃水浴回流1 h,然后加入磷酸二氢钾-氢氧化钠缓冲溶液(100 mL 0.2 mol/L磷酸二氢钾和69.5 mL 0.2 mol/L氢氧化钠混合)0.75 mL,混合液过减压ODS柱,先用35%甲醇洗脱10 mL,再用甲醇洗脱,收集甲醇洗脱液10 mL。用2.1.1项、2.2.1项的分析条件进行分析,得到如表7结果:
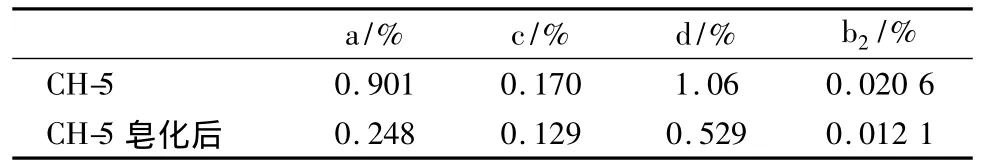
表7 皂化方法Ⅱ的结果Tab.7 Results of saponated actionⅡ
2.3.1.3 皂化方法Ⅱ的改进方法
药材粉末0.1 g,用90%甲醇提取3次,每次15 min,溶剂量分别为 2 mL,1.5 mL,1.5 mL,合并3 次滤液,用90%甲醇定容到5 mL,加入氢氧化钠溶液0.25 mL于50℃水浴回流1 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氢钾溶液50 mL洗涤,弃水相,再以40 mL水洗涤1次,有机相蒸干,精密加入10 mL甲醇定容。用2.1.1项、2.2.1项的分析条件进行分析,得到如表8结果:
对比以上3种方法,皂化方法Ⅱ以及皂化方法Ⅱ的改进方法都不能将柴胡中的皂苷完全的皂化,而皂化方法Ⅰ所得到的结果较为理想,所以选择皂化方法Ⅰ为实验方法。

表8 皂化方法Ⅱ改进方法的结果Tab.8 Results of improved saponated actionⅡ
2.3.2 供试品溶液的制备
精密称取柴胡药材约0.1 g,置于茄形瓶中,加5%氢氧化钾甲醇20 mL液浸渍过夜,加热回流5 h,提取液蒸干,加水30 mL,正丁醇溶液萃取(30 mL,30 mL,30 mL),合并萃取液加1%磷酸二氢钾溶液50 mL洗涤,弃水相,再以40 mL水洗涤1次,有机相蒸干,加甲醇溶解,定容10 mL,即为供试品溶液。
2.3.3 对照品溶液的制备
见2.1.4.1项,2.2.2项。
2.3.4 含量测定
将柴胡的各个批次的药材制备成供试品,按照2.1.1项、2.2.1项分析条件进行分析,结果见表9。
2.3.5 皂化反应前后总皂苷的含量测定,分析方法参考文献[6,7],方法学考察略。
皂化反应后,柴胡皂苷a、c、d以及a+c+d的含量在皂化反应前后变化幅度不大,但是柴胡皂苷b2的含量却大幅增加,增加了185%,而总皂苷的含量在皂化后却有所减少,减少了17.2%;醋炙柴胡、醋拌柴胡,在皂化反应后柴胡皂苷a、c、d以及a+c+d的含量变化并不大,大部分都有所减少,柴胡皂苷b2的含量几乎都有所增加,增加的幅度从24.6%~95.9%不等。总皂苷的含量也是增减不一,说明炮制品总皂苷的含量在皂化反应后无明显变化;对照药材,在皂化反应后,柴胡皂苷a+c+d的含量略微有所减少,但是柴胡皂苷b2的含量却大幅的增加,增加了117%,而总皂苷的含量在皂化反应后却有所减少,减少了49.9%;对照药材的炮制品醋拌柴胡,柴胡皂苷a、c、d以及a+c+d的含量在皂化反应后大幅度的减少,减少了72.2%,柴胡皂苷b2的含量也大幅度减少,减少了71.5%,总皂苷的含量也减少了30.4%。见表10。
3 讨论与结论
本实验首次采用皂化反应测定柴胡炮制前后皂苷的含量变化,前人研究表明柴胡含有大量的乙酰化皂苷成分[8],仅以柴胡皂苷 a、c、d、b2、b1作为对照,其结果有一定偏差,不能完全代表药材的实际含量,通过皂化反应去乙酰化后,分析结果较全面。在文献的基础上确定柴胡皂化反应方法,该法操作简单、重现性好、精密度良好,可以用于柴胡及其炮制品的皂苷成分含量分析。
通过结果说明,炮制后,总皂苷的含量都有所下降,柴胡皂苷b2的含量都大幅增加,柴胡皂苷a、c、d以及a+c+d的含量并没有什么变化。说明柴胡中的原生皂苷柴胡皂苷a、c、d在植物中的酸性成分条件下转化为次生苷柴胡皂苷b2、b1时,接近一半的柴胡皂苷b2、b1上面的羟基被乙酰化,而皂化反应后,乙酰基脱去,又重新转化为柴胡皂苷b2,所以含量大幅增加。而柴胡的炮制品中,可能被乙酰化的柴胡皂苷b2部分被去乙酰化,但并不完全,所以柴胡皂苷b2增加的幅度并没有柴胡大。以上的研究初步揭示了柴胡炮制后皂苷类化合物变化规律,为柴胡饮片的炮制及质控提供较为全面的参考。
柴胡炮制前后化学成分的比较分析,显示醋炙、醋拌这两种炮制工艺对柴胡总皂苷的含量变化没有显著影响,醋炙、醋拌是否可以通用,还需对其炮制后化学成分组成及其量比关系的变化进一步研究。

表9 柴胡各个样品皂化后皂苷的含量Tab.9 Results of saponated action

表10 柴胡各个样品皂化后总皂苷的含量Tab.10 Content of total saponins after saponated action
[1]中国药典[S].一部.2005:198.
[2]叶定江,原思通.中药炮制学辞典[M].上海:上海科学技术出版社,2005:295-296.
[3]刘 伟,黄国理.柴胡的不同炮制方法对其有效成分影响的研究[J].中药材,2005,18(1):21-23.
[4]李玲玲,张水龙.黄芪中有效成分环黄芪醇皂甙和黄芪甲甙的含量测定[J].中国现代应用药学,1998,15(3):13-15.
[5]Suzuki H,Yokota Y,Terasaki S,et al.Component Determination of Total Saponins in Bupleurum Root by High-performance Liquid Chromatography[J].Nat Med,2004,58(4):138-144.
[6]浦锦宝,胡轶娟,佘 靖,等.紫外可见分光光度法测定柴胡中柴胡总皂苷的含量[J].医学研究杂志,2008,37(6):98-100.
[7]郑虎占,董泽宏,佘 靖.中药现代研究与应用(第四卷)[M].北京:学苑出版社,1998:3688-3689.
[8]刘沁舡,谭 利,白焱晶,等.柴胡属植物皂苷近10年研究概况[J].中国中药杂志,2002,27(1):7-11.
猜你喜欢
石油与天然气化工(2023年6期)2023-12-31 04:02:32
江南诗(2023年6期)2023-12-08 05:17:24
遵义医科大学学报(2023年1期)2023-02-06 02:18:40
金沙江文艺(2022年1期)2022-02-04 10:15:16
化学教与学(2021年12期)2021-02-18 01:16:58
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24 21:02:17
中成药(2018年4期)2018-04-26 07:13:09
中成药(2017年8期)2017-11-22 03:18:49
安阳工学院学报(2016年6期)2016-12-06 02:20:06
特产研究(2015年2期)2015-03-24 06:17:43
