
重组丙型肝炎病毒核心蛋白的原核表达、纯化和抗体制备
2010-01-26 06:38陈辉高娜范东瀛郑群王娟安静
微生物与感染 2010年3期
陈辉,高娜,范东瀛,郑群,王娟,安静
首都医科大学基础医学院微生物学教研室,北京 100069
丙型肝炎病毒(hepatitis C virus,HCV) 是导致慢性肝炎、肝纤维化甚至肝细胞癌的重要原因之一,全世界感染人数已超过2.27亿,其中中国感染人数4 000万, HCV感染已成为全球化的健康难题[1]。HCV相关肝细胞癌的分子机制目前尚不十分清楚,由于缺乏特效治疗手段和安全有效的疫苗,所以对其致病机制的研究已成为HCV 感染亟待解决的前沿课题之一。
HCV属黄病毒科(Flaviviridae),根据5′非编码区序列的差异可将不同HCV株分为6个基因型和100多个基因亚型,其中中国内地地区HCV以1b亚型为主。HCV基因组为单正链RNA,编码3 010个氨基酸组成的大分子蛋白单体,包括3种结构蛋白以及至少7种非结构蛋白。其中HCV核心蛋白(HCV core protein, HCV-C) 为由191个氨基酸残基组成的前体蛋白在细胞内进一步加工形成的174个氨基酸多肽片段,是与RNA结合的亲膜α螺旋蛋白二聚体,参与病毒核衣壳的形成,是HCV基因组中较为保守的结构。更重要的是,HCV-C能与宿主细胞蛋白相互作用,进而影响信号转导、细胞凋亡、肿瘤发生及脂质代谢,参与丙型肝炎的发生和发展[2]。特别是HCV-C具有反式激活作用,是导致病毒持续性感染以及肝细胞癌发生的重要原因,因而备受关注[3],但其致病机制有待进一步阐明。本研究旨在获得纯化的HCV-C,制备抗HCV-C的多克隆抗体,为深入研究HCV-C与肝细胞相互作用的分子机制奠定基础。
1 材料和方法
1.1 菌株、细胞、质粒和动物
大肠埃希菌JM109和Vero细胞由本实验室保存。模板质粒HCV1b亚型HC-J4全基因组由第二军医大学戚中田教授惠赠。原核表达载体pQE31购自Promega公司。真核表达重组质粒pCAGGSP7-HCV-C由本室保存。6~8周龄雌性BALB/c小鼠由军事医学科学院实验动物中心提供。
1.2 主要试剂
镍亲和层析柱购自Invitrogen公司。聚合酶链反应(polymerase chain reaction,PCR)试剂为TaKaRa公司产品。DNA限制性内切酶PstⅠ、HindⅢ以及T4连接酶均购自MBI公司。质粒抽提试剂盒购自北京博大泰克基因技术公司。DNA凝胶回收试剂盒购自Qiagen公司。DNA相对分子质量标准品与蛋白质相对分子质量标准品购自Fermentas公司。弗氏佐剂购自Gibco BRL公司。转染试剂Lipofectamine 2000为Invitrogen公司产品。小鼠抗HCV-C单克隆抗体(产品编号ab2740)购自Abcam公司。羊抗小鼠IgG-FITC、IgG-HRP购自Sigma公司。其他常规化学试剂均为分析纯产品。
1.3 引物设计和目标片段的PCR扩增
根据HCV1b亚型HC-J4-91(GenBank序列号:AF054250)核心蛋白的核苷酸序列,采用Primer Premier 5.0软件,设计合成特异性引物(上海英骏生物技术有限公司)。pQE31-HCV-C上游引物:5′- GGCACTGCAGATGAGCACGAATCCTAA ACC-3′;下游引物:5′-GTGAAGCTTTTAAGCGGAAGCTGGGAT -3′。上游引物含酶切保护性碱基及PstⅠ酶切位点(单下划实线处),下游引物含终止密码子TTA(双下划实线处)、HindⅢ酶切位点(单下划虚线处)以及酶切保护性碱基。以HCV1b亚型HC-J4-91全基因组质粒为模板,PCR扩增目的基因片段。反应参数:在50 μl反应体积中依次加入5×PrimeSTARTMbuffer(Mg2+plus) 10 μl,2.5 mmol/L dNTP mixture 4 μl,25 pmol/μl的上、下游引物各1 μl,cDNA模板1 μl,PrimeSTARTMHS DNA polymerase 1 μl,补加双蒸水至50 μl,混匀后进行扩增。扩增步骤:95 ℃预变性5 min;98 ℃ 10 s,58 ℃15 s,72 ℃ 1 min,30个循环;72 ℃延伸10 min。取PCR产物于10 g/L琼脂糖凝胶电泳,紫外分析仪检测电泳结果。
1.4 pQE31-HCV-C表达质粒的构建
从琼脂糖凝胶回收纯化PCR产物中572 bp的目的基因片段,PstⅠ、HindⅢ双酶切后,以T4连接酶于16 ℃反应16 h,连接同样双酶切的载体pQE31,构建原核表达重组质粒pQE31-HCV-C。
1.5 重组质粒转化大肠埃希菌及测序鉴定
将连接产物化学转化感受态JM109工程菌,挑取转化菌落扩增培养,提取质粒,经PCR及PstⅠ、HindⅢ双酶切鉴定无误后,将含目标序列的重组质粒工程菌送大连宝生物工程有限公司测序鉴定。
1.6 融合蛋白6×His-HCV-C诱导表达条件的优化
1.6.1融合蛋白6×His-HCV-C的异丙基硫代半乳糖苷最佳诱导浓度的确定
将含pQE31-HCV-C质粒和pQE31空质粒的工程菌分别接种于含100 μg/ml氨苄西林的2×YT培养基中,37 ℃,160 r/min,培养过夜;次日以1∶100接种于新鲜培养基,培养至菌液A600约0.6,加入异丙基硫代半乳糖苷(isopropyl β-D-1-thiogalactopyranoside,IPTG)至终浓度分别为0.2、0.4、0.6、0.8、1.0 mmol/L,诱导表达6 h后收获菌体,进行十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)分析,积层胶50 g/L,分离胶100 g/L,100 V、15 min后再稳压130 V电泳40 min。
1.6.2融合蛋白6×His-HCV-C的IPTG最佳诱导时间的确定
将含pQE31-HCV-C质粒和pQE31空质粒的工程菌分别接种于含100 μg/ml氨苄西林的2×YT培养基中,37 ℃,160 r/min,培养过夜;次日以1∶100接种于新鲜培养基,培养至菌液A600约0.6,加入IPTG至终浓度为0.6 mmol/L,分别诱导表达0、1、2、3、4、5、6 h后收获菌体,进行SDS-PAGE分析,积层胶50 g/L,分离胶100 g/L,电压100 V、15 min后再稳压130 V电泳40 min。
1.7 融合蛋白6×His-HCV-C的纯化
800 ml菌液经离心,收集表达后所获的菌体,以Buffer A(50 mmol/L Tris pH 8.0、0.5 mmol/L EDTA、50 mmol/L NaC1、50 ml/L甘油)洗涤2次并重悬,加入1 mmol/L苯甲基磺酰氟(phenylmethanesulfonyl fluoride,PMSF)及1 mg/ml溶菌酶,超声破碎后加1 ml/L Triton X-100,4 ℃静置30 min后,12 000g离心1 h,收集沉淀物,Buffer A洗涤沉淀2次,加入Buffer B(8 mol/L尿素、100 mmol/L NaH2PO4、1 ml/L巯基乙醇、10 mmol/L Tris pH 8.0),于4 ℃溶解包涵体沉淀。样品通过镍亲和层析柱,用Buffer C(8 mol/L尿素、100 mmol/L NaH2PO4、10 mmol/L Tris pH 6.3)去除杂蛋白,Buffer D(8 mol/L尿素、100 mmol/L NaH2PO4、10 mmol/L Tris pH4.5)洗脱目的蛋白。
经透析去除尿素,获得纯化的HCV-C复性蛋白。利用SDS-PAGE检测6×His-HCV-C融合蛋白的纯度。
1.8 抗HCV-C多克隆抗体的制备
将1 ml 200 μg/ml目的蛋白与等体积弗氏佐剂充分混匀,皮下、腹腔及肌肉多点注射BALB/c小鼠,每隔3周加强免疫1次,共免疫3次。末次免疫1周后采血,分离血清,间接酶联免疫吸附试验(enzyme-linked immunosorbent assay,ELISA)检测其抗体效价。
1.9 蛋白免疫印迹
为鉴定所制备抗体的特异性,将纯化融合蛋白经SDS-PAGE,80 V电泳2 h,转印至硝酸纤维素滤膜,用自制的小鼠抗HCV-C血清(1∶500 稀释)与膜4 ℃孵育过夜。以羊抗鼠IgG-HRP为二抗,二氨基联苯胺(3,3’-diaminobenzidine,DAB)显色观察,至目的条带清晰终止反应。
1.10 间接免疫荧光染色
制备Vero细胞的细胞爬片,提取真核表达质粒pCAGGSP7-HCV-C DNA,利用Lipofectamine 2000转染Vero细胞,48 h后取出,经40 ml/L聚合甲醛固定、2 ml/L Triton X-100/磷酸缓冲液(phosphate-buffered saline,PBS)通透后,以10 ml/L 牛血清白蛋白(bovine serum albumin,BSA)/PBS室温封闭1 h,加入制备的小鼠抗HCV-C血清(1∶200稀释)或市售小鼠抗HCV-C单克隆抗体(1:200稀释),4 ℃孵育过夜。漂洗后加入羊抗鼠IgG-FITC,室温孵育1 h,经漂洗、干燥后,用400 ml/L甘油封片,荧光显微镜(Olympus BX61)观察。
2 结果
2.1 HCV-C DNA片段的获得及克隆入载体pQE31
以HCV1b亚型HC-J4-91全基因组质粒为模板,利用上述特异性引物进行PCR扩增,将PCR产物于10 g/L琼脂糖电泳鉴定,可见500~750 bp特异性条带,与预期目的基因572 bp片段长度相符。将PstⅠ、HindⅢ双酶切目的基因片段连接于同样双酶切的载体pQE31并转化JM109工程菌,提取重组质粒经PCR及PstⅠ、HindⅢ双酶切鉴定,扩增产物与酶切产物中均含572 bp目的基因片段(图1)。重组质粒pQE31-HCV-C经测序,证实目的基因片段正确插入pQE31载体的单克隆位点,并保持正确的读码框架。
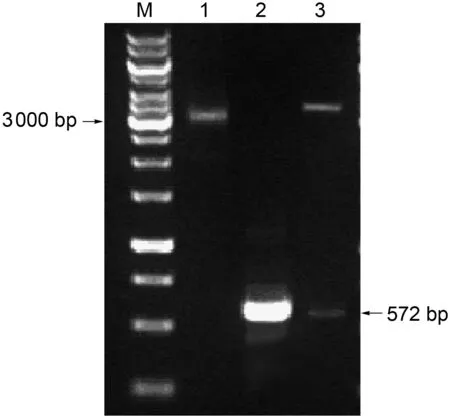
1,recombinant plasmids; 2, PCR amplification; 3,PstⅠ/HindⅢ digestion.
图1pQE31-HCV-C重组原核表达质粒酶切及PCR鉴定图谱
Fig.1RestrictionmapandPCRanalysisofpQE31-HCV-C
2.2 融合蛋白6×His-HCV-C 的原核表达
按照前述方法,将含pQE31-HCV-C质粒的重组工程菌以终浓度分别为0.2、0.4、0.6、0.8、1.0 mmol/L的IPTG诱导表达6 h后收获菌体,进行SDS-PAGE分析。结果表明,与空载体标本及诱导前标本相比,诱导标本中出现22 000的新生蛋白条带(图2)。融合蛋白主要以包涵体形式存在,其相对分子质量与预期相符,且IPTG终浓度为0.6 mmol/L时,诱导表达6 h目的蛋白表达量最高,故以此确定为融合蛋白6×His-HCV-C的IPTG最佳诱导条件。
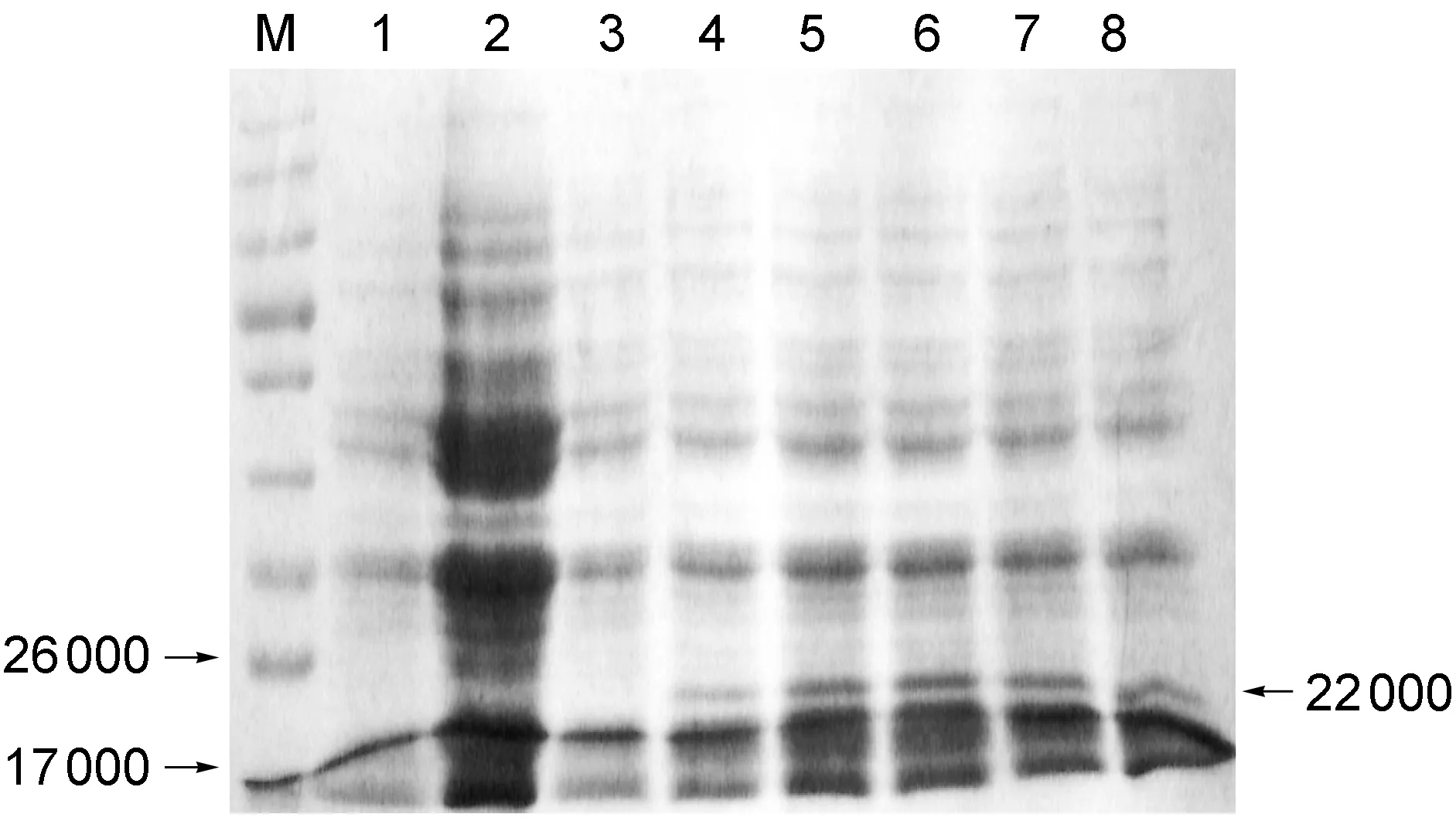
1 and 2, JM109/pQE31 induced by 1.0 mmol/L IPTG for 0 h and 6 h, respectively; 3-8, JM109/pQE31-HCV-C induced by 0, 0.2, 0.4, 0.6, 0.8, 1.0 mmol/L IPTG for 6 h.
图2SDS-PAGE分析不同浓度IPTG诱导融合蛋白6×His-HCV-C的原核表达
Fig.2SDS-PAGEanalysisoftheprokaryoticexpressionof6×His-HCV-CfusionproteininducedbyIPTGatdifferentconcentrations
2.3 融合蛋白6×His-HCV-C的纯化及纯度分析
将含pQE31-HCV-C质粒的重组工程菌以终浓度0.6 mmol/L的IPTG诱导表达6 h后收获菌体,超声破碎后菌体沉淀物经镍亲和层析获得纯化融合蛋白。蛋白样品经SDS-PAGE分析,蛋白相对分子质量约22 000。灰度扫描结果显示,纯化融合蛋白的纯度达90%(图3)。
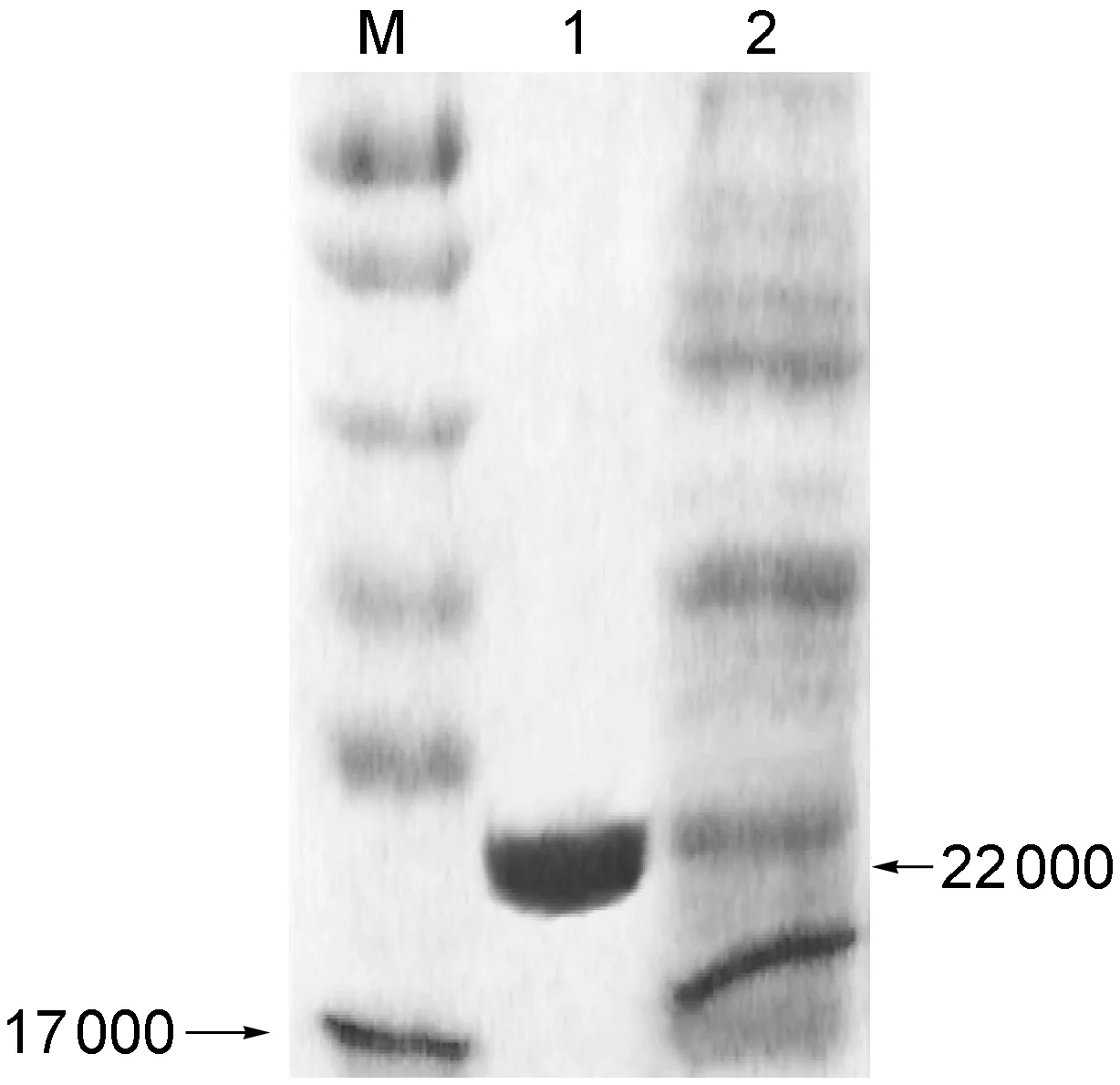
1, purified 6×His-HCV-C fusion protein; 2,E.coliJM109 strains expressing 6×His-HCV-C fusion protein induced by IPTG at 0.6 mmol/L for 6 h.
图3纯化后融合蛋白的SDS-PAGE电泳分析
Fig.3SDS-PAGEanalysisofthepurified6×His-HCV-Cfusionprotein
2.4 HCV-C多克隆抗体的制备及检测
小鼠抗血清经间接ELISA检测,抗体效价达1∶12 800。蛋白免疫印迹检测结果证明,1∶500 稀释的自制小鼠抗HCV-C多克隆抗体能够与诱导表达的6×His- HCV-C发生特异的抗原抗体反应(图4)。
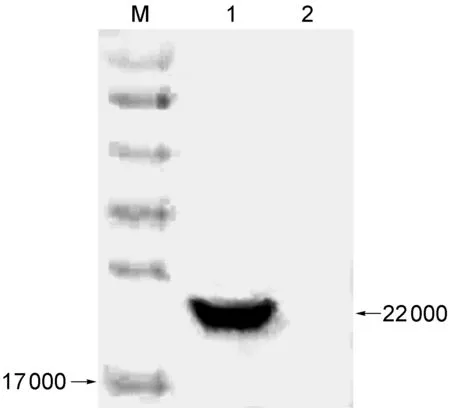
1, polyclonal antibodies against HCV-C; 2, normal mouse serum.
图4蛋白免疫印迹鉴定小鼠抗HCV-C抗血清
Fig.4IdentificationofpolyclonalantibodiesagainstHCV-CbyWesternblot
2.5 间接免疫荧光染色
将真核表达质粒pCAGGSP7-HCV-C转染Vero细胞,48 h后取出,利用自制小鼠抗血清作为一抗进行染色,观察HCV-C的表达情况。结果显示,Vero细胞胞质内出现明显的特异性荧光,与市售小鼠抗HCV-C单克隆抗体分布模式一致,表明自制小鼠抗HCV-C多克隆抗体可特异性识别真核表达的HCV-C(图5),而正常血清对照组无特异性荧光。
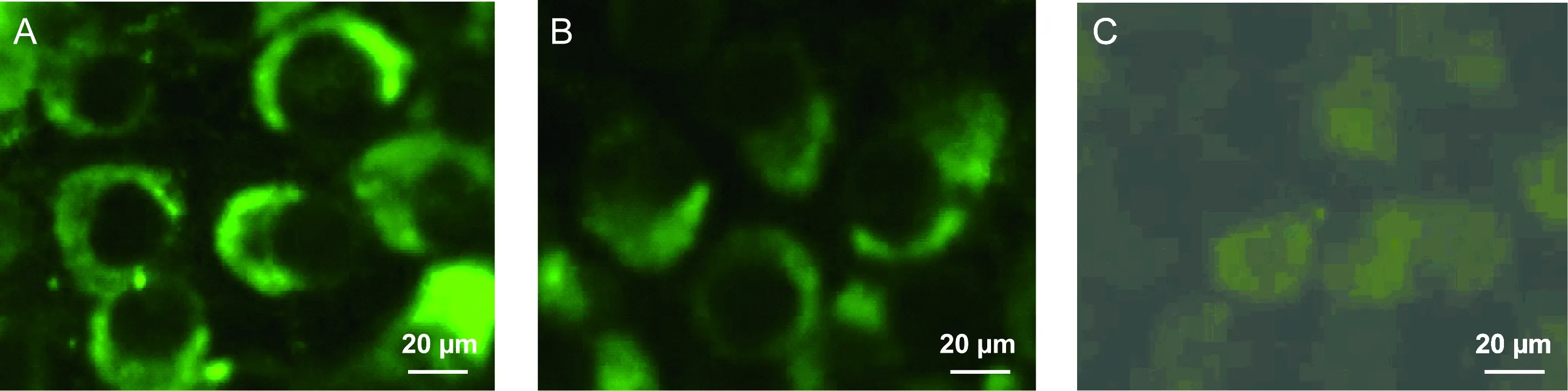
A: The polyclonal antibodies against HCV-C. B: Commercial monoclonal antibodies against HCV-C. C: Normal mouse serum.
图5间接免疫荧光染色检测HCV-C在Vero细胞中的表达
Fig.5DetectionofHCV-CexpressioninVerocellswithantibodiesagainstHCV-Cbyindirectimmunofluorescencestaining
3 讨论
在HCV编码的结构蛋白和非结构蛋白中,HCV-C的功能最复杂,除在病毒颗粒装配与成熟过程中起关键作用外[4],还参与调节细胞凋亡、脂代谢、病毒基因表达及抗原呈递等。HCV-C与宿主细胞的多种蛋白相互作用可能是HCV慢性感染,肝细胞损伤以至肝细胞癌变发生、发展的主要成因[5]。有研究认为,HCV感染后,HCV-C等可通过免疫病理反应造成肝细胞损伤,诱导肝细胞凋亡。凋亡的肝细胞可刺激单核-巨噬细胞等释放白细胞介素1β(interleukin 1β,IL-1β)、肿瘤坏死因子α(tumor necrosis factor α,TNF α)等大量炎性细胞因子,影响肝细胞内信号转导通路,最终可能导致肝癌发生[6,7]。研究证明,HCV-C前体在内质网上被信号肽酶切掉C端疏水区后,加工为成熟的HCV-C,可与核蛋白酶活化分子PA28γ联合作用,致使肝细胞变性、坏死[8]。这种作用可能与HCV-C诱导肝组织中活性氧的表达有关。此外,HCV-C可作用于肿瘤相关的重要信号通路,抑制肝癌细胞凋亡,促进肝细胞增殖并转化。Herzer等[9]发现,HCV-C可通过与P53蛋白的共活化分子相互作用,阻止抑癌基因p53激活。Battaglia等[10]研究指出,HCV-C能抑制转化生长因子β(transforming growth factor β,TGF β)介导的抑癌通路,导致细胞增殖的失调。
HCV-C也是HCV与宿主免疫细胞相互作用的主要环节,在HCV感染后的免疫逃逸中发挥重要作用[11]。Zimmermann等[12]用表达HCV-C的小鼠肝细胞进行体外实验,发现HCV-C可通过下调抗原呈递细胞的组织相容性复合物(major histocompatibility complex,MHC)Ⅰ类分子、α干扰素(interferon α,IFN α)和TNF α表达的同时,诱导调控性细胞因子IL-10的生成,从而抑制CD8+T细胞免疫反应起始,造成HCV在体内不易被清除而呈长期慢性感染。此外,HCV-C还可通过上调无反应基因和新异基因的表达影响T细胞的内环境,造成机体免疫功能障碍[13]。由此可见HCV-C在HCV感染与致病中具有重要作用,但机制较为复杂,尚未完全阐明。
为深入研究HCV-C 与肝细胞相互作用的分子机制,本实验构建了HCV-C的原核表达质粒,进而获得其纯化蛋白并制备了抗HCV-C的多克隆抗体。实验中采用的pQE31原核表达载体带有6个连续组氨酸残基序列,与目的基因片段共同表达后,组氨酸标签可与镍螯合,使目的蛋白被特异性吸附于亲和层析柱;再利用pH梯度分段洗脱,达到分离、纯化蛋白的目的。结果表明,该方法获得的纯化融合蛋白相对分子质量约22 000,纯度较高,可满足实验要求。利用此重组蛋白免疫小鼠,所制备的抗血清效价较高,并能特异性识别自然状态(免疫荧光染色)和变性的(蛋白免疫印迹)HCV-C,说明该抗血清可用于ELISA、蛋白免疫印迹以及免疫荧光染色等实验研究。进一步以市售单克隆抗体为对照,进行免疫荧光染色,发现与自制抗体的特异性染色分布方式一致,说明所制备的抗体有较好的实用性。该制备方法简单,所获抗体有实用价值,克服了市售抗体价格昂贵的缺点,为进一步研究HCV-C与肝细胞相互作用的分子机制提供了重要的实验工具。
[1] Bartosch B, Thimme R, Hubert E, Blum, Zoulim F. Hepatitis C virus-induced hepatocarcinogenesis [J]. J Hepatol, 2009, 51(4): 810-820.
[2] Irshad M, Dhar I. Hepatitis C virus core protein: an update on its molecular biology, cellular functions and clinical implications [J]. Med Princ Pract, 2006, 15(6): 405- 416.
[3] Tran G. The role of hepatitis C virus in the pathogenesis of hepatocellular carcinoma [J]. Res Horiz, 2008, 1 (2): 167-175.
[4] Klein K C, Delloss S R, Lingappa J R. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell free system [J]. J Virol, 2005, 79(11): 6814-6826.
[5] Dhumeaux D. Steatosis, fibrosis and hepatitis C virus infection [J]. Arab J Gastroenterol, 2010, 10(4 Suppl): S30-S31.
[6] Saito K, Meyer K, Warner R, Basu A, Ray RB, Ray R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein [J].J Virol, 2006, 80(9): 4372- 4379.
[7] Spaziani A, Alisi A, Sanna D, Balsano C. Role of p38 MAPK and RNA-dependent protein kinase (PKR) in hepatitis C virus core-dependent nuclear delocalization of cyclin B1 [J]. J Biol Chem, 2006, 281(16): 10983-10989.
[8] Mori Y, Moriishi K, Matsuura Y. Hepatitis C virus core protein: Its coordinate roles with PA28 gamma in metabolic abnormality and carcinogenicity in the liver [J]. Int J Biochem Cell Biol, 2008, 40(8): 1437-1442.
[9] Herzer K, Weyer S, Krammer PH, Galle PR, Hofmann TG. Hepatitis C virus core protein inhibits tumor suppressor protein promyelocytic leukemia function in human hepatoma cells [J]. Cancer Res, 2005, 65(23): 10830-10837.
[10] Battaglia S, Benzoubir N, Nobilet S, Charneau P, Samuel D, Zignego AL, Atfi A, Christian B, Bourgeade MF. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition [J]. PLoS ONE, 2009, 4(2): e4355.
[11] Szabo G, Dolganiuc A. Hepatitis C core protein—The “core” of immune deception [J]? J Hepatol, 2008, 48(1): 8-11.
[12] Zimmermann M, Flechsig C, Lamonica N. Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes [J]. J Hepatol, 2008, 48(1): 51-60.
[13] Domonguez VM, Munoz SA, Anaya BB, Aguilar S, Novalbos JP, Giron JA, Rodriguez IM, Garcia CF. Hepatitis C virus core protein up regulates anergy-related genes and a new set of genes, which affects T cell homeostasis [J]. J Leukoc Biol, 2007, 82(5): 1301-1310.
猜你喜欢
传染病信息(2022年6期)2023-01-12
昆明医科大学学报(2022年2期)2022-03-29
江西农业学报(2021年4期)2021-04-20
江苏农业科学(2019年9期)2019-08-20
湖北农业科学(2017年8期)2017-05-26
广东饲料(2016年1期)2016-12-01
大陆桥视野·下(2016年6期)2016-08-06
癌变·畸变·突变(2016年3期)2016-02-27
哈尔滨医药(2015年4期)2015-12-01
西南医科大学学报(2015年1期)2015-08-22
