
铁死亡抑制基因在食管癌中的高表达分析
2024-11-03 00:00:00王元国张鹏
南方医科大学学报 2024年7期
关键词:细胞凋亡


摘要:目的 通过生物信息学分析和实验验证探讨铁死亡基因在食管鳞癌中的作用。方法 从基因表达数据库(GEO)下载食管鳞癌数据集GSE161533和GSE20347,采用R软件分析获得差异基因(DEGs),将DEGs与铁死亡数据库(FerrDb)中的基因取交集获得食管癌铁死亡相关基因,将获得食管癌铁死亡相关基因进行基因本体论(GO)和京都基因与基因组百科全书(KEGG)分析、String构建蛋白互作网络(PPI)、Cytoscape筛选铁死亡核心基因及抑制基因,筛选获得食管癌铁死亡抑制基因在癌症基因图谱(TCGA)中进行验证。应用qPCR检测食管癌铁死亡抑制基因在正常食管细胞和食管癌细胞中的表达水平,在TE1食管癌细胞系中下调6个食管癌铁死亡抑制基因,分为对照组(Control组)、siRNA阴性对照组(Negative组)和每个siRNA干扰组(包括RRM2-siRNA组、GCLC-siRNA组、TFRC-siRNA组、TXN-siRNA组、SLC7A11-siRNA组和EZH2-siRNA组),对照组不加任何转染试剂,其余各组使用Lipofectamine 2000(终浓度为3 μL/mL)和siRNA(终浓度为 100 nmol/L)的转染体系转染细胞,转染后分别进行流式细胞学实验、CCK8实验以验证食管癌铁死亡抑制基因对食管癌细胞功能的影响,Western blotting实验验证食管癌铁死亡抑制基因对铁死亡进程的影响。结果 共获得58个食管癌铁死亡相关基因,GO分析显示其主要参与谷胱甘肽跨膜转运、铁离子输运和细胞凋亡等生物学过程,KEGG富集分析显示其主要参与铁死亡、谷胱甘肽代谢、抗叶酸抵抗等信号通路。PPI 网络包含54 个节点和74 条边,局部聚类系数为0.522,PPI 富集值为Plt;0.001。RRM2、TFRC、TXN、EZH2、SLC7A11、GCLC 6个食管癌铁死亡抑制基因,在TCGA数据集的食管癌组织中表达高于正常食管组织(Plt;0.01),与正常食管细胞系比较,这些基因在食管癌细胞系中表达升高(Plt;0.05)。在食管癌TE1 细胞中,应用siRNA分别下调RRM2、TFRC、TXN、EZH2、SLC7A11、GCLC可抑制细胞增殖(Plt;0.05)、促进细胞凋亡(Plt;0.05),铁死亡进程标志蛋白GPX4、FIH1 表达下调(Plt;0.05)、ACSL4表达上调(Plt;0.05)。结论 铁死亡抑制基因在食管癌中的高表达可能阻碍了食管癌细胞的铁死亡进程,导致肿瘤发生,抑制这些基因可恢复铁死亡进程,促进细胞凋亡,铁死亡抑制基因有望成为食管癌治疗的新靶点。
关键词:食管鳞癌;铁死亡;细胞凋亡
食管癌是一种常见的恶性度高的消化系统肿瘤,其早期症状隐匿,患者确诊时多处于晚期,其5 年生存率不到30%[1] 。当前,食管癌的治疗主要依赖于手术、放疗和化疗,虽然免疫治疗和靶向治疗显示出一定的临床潜力[2] ,但由于有效的靶向治疗药物较少,整体治疗效果仍不理想。近年来,高通量测序技术及生物信息学在肿瘤医学中的应用日益广泛,这些先进技术帮助研究者揭示了肿瘤发生发展的分子机制,并筛选出用于早期诊断、预后评估和指导个体化治疗的潜在肿瘤生物标志物。因此,应用基因测序和生物信息技术寻找食管癌发病的基因组特征,并筛选相关靶点,对揭示其发病机制和开发靶向治疗药物具有重要的意义。
铁死亡作为一种依赖于铁和活性氧(ROS)积累的细胞死亡方式,近年来在肿瘤研究中倍受关注[3-5]。铁死亡与传统的细胞凋亡、坏死和自噬不同,其主要特征为细胞内铁离子过量积累和脂质过氧化,最终导致细胞膜的损伤和细胞死亡。越来越多的研究表明,在乳腺癌、卵巢癌、肺癌等多种肿瘤中,通过诱导肿瘤细胞铁死亡,可抑制肿瘤生长[3, 4]。目前,已有多种铁死亡诱导剂被开发并用于肿瘤治疗,这些诱导剂主要通过抑制半胱氨酸摄取和谷胱甘肽过氧化物酶4(GPX4)合成、诱导脂质过氧化,促进肿瘤细胞的铁死亡[5, 6]。食管癌的发病机制涉及多个复杂的生物学过程,包括基因组变异、肿瘤抑制基因的失活、癌基因的异常活化和氧化应激等。GPX4在铁死亡中起着重要调节作用,其主要机制是通过清除细胞膜上的脂质过氧化物,增强细胞对氧化应激的抵抗能力,抑制铁死亡的发生[7, 8]。研究表明,食管鳞癌组织中GPX4表达上调,说明铁死亡被抑制,而下调食管癌细胞的GPX4表达可诱导铁死亡信号通路活化,发挥抗肿瘤作用[9, 10]。尽管已有研究表明,铁死亡参与了食管癌的发生和转移[11],但其具体的调控机制仍不明确,相关研究仍然较少。环状RNA PVT1是食管癌发病的重要调节因子,可通过miR-30a-5p/FZD3通路调节化疗敏感性,PVT1在5-氟尿嘧啶耐药的食管癌细胞中显著上调,敲低PVT1可明显降低GPX4和SLC7A11的表达水平,诱导细胞铁死亡[12]。青蒿琥酯、柳氮磺吡啶、萜类和砷类等化合物可以通过直接或间接地诱导铁死亡,抑制食管癌细胞的生长和迁移[13-16]。虽然这些研究提示铁死亡机制参与了食管癌的发生发展,但铁死亡基因在食管癌中的表达和意义尚不清楚。
因此,本研究拟从基因表达综合数据库(GEO)下载食管鳞癌相关的数据集,筛选出差异表达基因(DEGs),并与铁死亡基因数据集取交集后进行生物信息学分析,筛选得到具有重要意义的食管癌铁死亡基因并进行实验验证,为食管癌中铁死亡机制的研究提供理论依据和研究思路,以期为食管癌的治疗提供潜在靶点。
1 材料和方法
1.1 基因表达谱数据集筛选
登录NCBI 网站,选择GEO数据库(https://www.ncbi.nlm.nih.gov/geo/),在GEO Datasets检索框中输入“Esophageal squamous cell carcinoma”进行检索,且每个数据集总样本数不低于30例,最终获得GSE161533和GSE20347两个数据集。GSE161533包括28个食管癌组织,28 个配对的正常食管组织;GSE20347 包括包括17个食管癌组织,17个配对的正常食管组织。两个数据集均基于Affymetrix Human Genome U133 Plus2.0 Array平台。
1.2 食管鳞癌DEGs的筛选
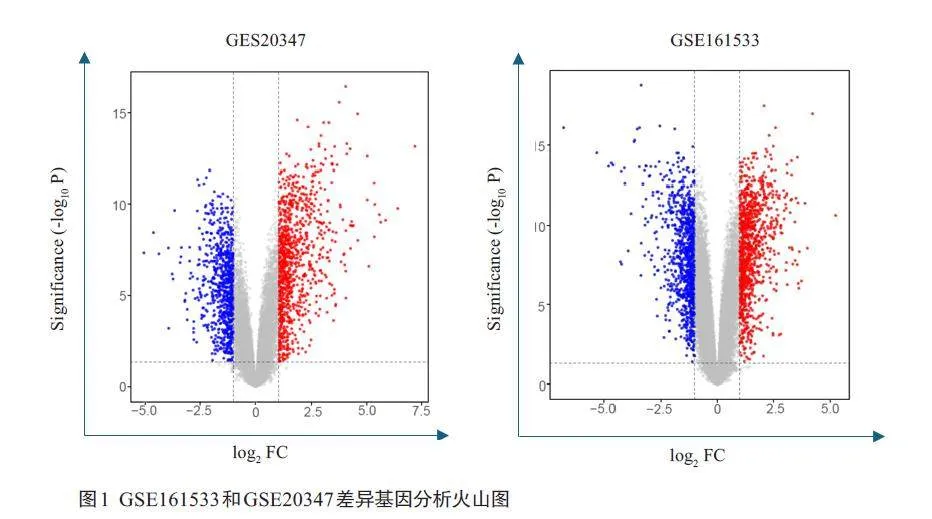
在GEO网站下载2 个数据集的数据和平台文件,根据样本编号分为食管癌组和正常食管组,使用R语言将探针名注释为基因名,基因名重复的表达谱数据取均值,删除未检测到数据(NA)及空值,使用R 软件的“limma”包进行差异基因筛选,筛选标准为校正Plt;0.05,|logFC|≥1,获得筛选的DEGs 后,以-log10(P)为纵坐标,以log2FC为横坐标,分别构建火山图。
1.3 食管鳞癌与铁死亡相关基因筛选
通过FerrDb V2 数据库,检索铁死亡“Driver”“Suppressor”“Marker”相关基因,将铁死亡相关基因与2个数据集的食管鳞癌DEGs取交集,绘制韦恩图,交集即为食管癌与铁死亡相关基因。
1.4 GO、KEGG富集和PPI分析
应用R软件“ClusterProfiler”包对食管癌与铁死亡相关基因进行基因本体论(GO)功能和京都基因与基因组百科全书(KEGG)信号通路富集分析分析,GO分析包括的生物学过程(BP)、分子功能(MF)和细胞组分(CC)分析。数据过滤条件为P value Cutoff=0.05,qvalue Cutoff=0.05,获得富集数据后,应用“ggplot2”包对富集的数据进行可视化分析。
将食管癌与铁死亡相关基因导入STRING数据库(https://cn. string-db. org/),进行蛋白-蛋白互作网络(PPI)分析,互作评分gt;0.4,STRING分析后,将得到的数据导入Cytoscape 软件,删除孤立的节点,计算节点和边,应用Network 模块对蛋白-蛋白互作数据进行可视化分析。
1.5 食管癌铁死亡抑制基因筛选及TCGA验证
在Cytoscape 中,应用“MCC”、“Degree”和“Closeness”3种统计方法分别筛选出前10位的食管癌铁死亡基因,取交集后获得核心基因,并将核心基因与铁死亡抑制基因取交集,获取食管癌铁死亡抑制基因。
下载TCGA数据集中的食管癌数据,从中筛选食管鳞癌和正常食管组织中食管癌铁死亡抑制基因的相关测序数据,比较这些基因在两组之间的表达差异。
1.6 细胞系验证食管癌铁死亡抑制基因表达水平
复苏本实验室留存的人正常食管上皮细胞系(HEEC)和人食管鳞癌细胞系(TE1 和KYSE150),HEEC应用含10%胎牛血清的DMEM高糖培养基培养,2 株食管癌细胞系均使用含10% 胎牛血清的RMPI 1640培养基培养。细胞稳定传代2次后,各组取106个细胞提取RNA,应用逆转录试剂盒将1 μg RNA逆转录为cDNA,稀释5倍后使用。委托上海生工合成扩增引物,引物序列见表1,应用荧光定量试剂盒检测基因表达水平,在扩增体系中加入上游引物2 μL,下游引物2 μL,cDNA 1 μL,TB Green 10 μL,ddH2O 5 μL,离心混匀后置qPCR仪进行基因扩增,扩增条件为:95 ℃30 s,56 ℃ 30 s,72 ℃ 20 s,40cycles。应用2-ΔΔCt法计算食管癌铁死亡抑制基因mRNA的相对表达水平。
1.7 下调铁死亡抑制基因对食管癌细胞凋亡、增殖和铁死亡的影响
1.7.1 试剂
TE1食管癌细胞系(本实验室留存);GPX4抗体、FIH1 抗体、SCSL3 抗体、GAPDH抗体(Abcam);羊抗兔二抗和羊抗小鼠二抗(Proteintech);ECL发光液(Advansta);Silencer Select siRNA:RRM2 siRNA、GCLC siRNA、TFRC siRNA、TXN siRNA、SLC7A11siRNA、EZH2 siRNA、Negative siRNA(Thermo FisherScientific);Opti-MEM培养基、RMPI1640培养基、胎牛血清(Gibico);Lipofectamine 2000(Invitrogen);细胞计数试剂盒-8(CCK-8)、Annexin V-FITC/7AADPercpCy5.5(普诺赛)。
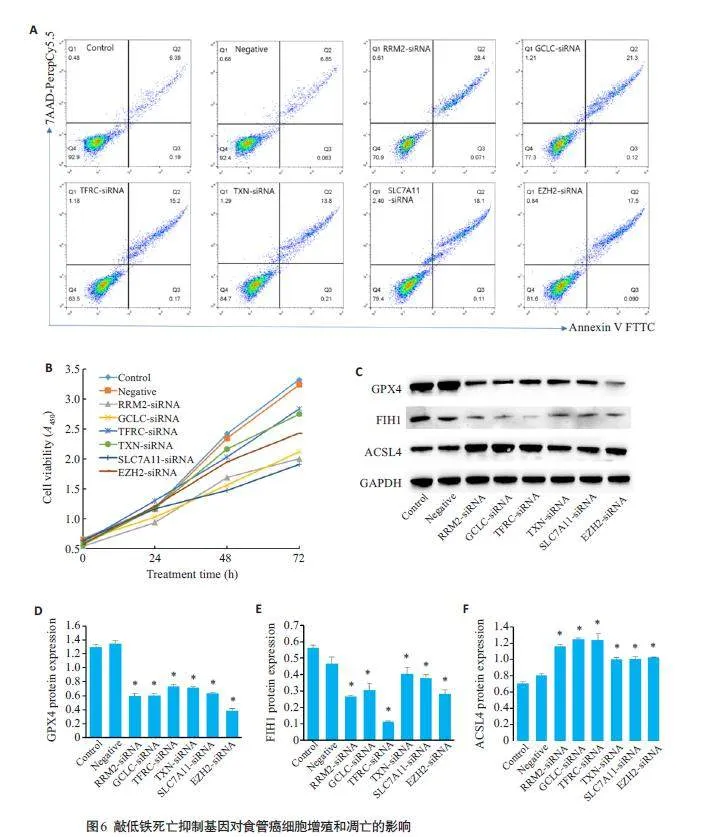
1.7.2 细胞增殖能力
TE1 细胞稳定传代3 次后进行siRNA干扰实验。胰蛋白酶消化TE1细胞,调整细胞密度为20 000/mL,向96 孔板的每孔加入100 μL细胞悬液,培养至细胞融合度至40%,将细胞分为对照(Control组)、siRNA 阴性对照组(Negative 组)、RRM2 siRNA组、GCLC siRNA组、TFRC siRNA组、TXN siRNA组、SLC7A11 siRNA组、EZH2 siRNA组,每组3个复孔,转染前1 d更换含10%胎牛血清且无抗生素的Opti-MEM培养基。转染当天Control 组仅加100 μL Opti-MEM培养基,其余各组分别加入Lipofectamine 2000(终浓度为3 μL/mL)、各组siRNA(终浓度为 100 nmol/L)和Opti-MEM培养基至100 μL,转染6 h后更换含10%胎牛血清的RMPI 1640培养基,分别于第0、24、48、72 h向各孔内加入10 μL CCK-8溶液,细胞培养箱孵育3 h后,应用酶标仪测定各孔吸光度A450 nm。
1.7.3 细胞凋亡
转染体系和时间同1.7.2,siRNA 转染48 h后应用胰蛋白酶消化细胞,获得单细胞悬液,应用1×PBS洗涤1次,300 g离心5 min,弃上清后加AnnexinV-FITC/7AAD-PercpCy5.5试剂盒中的400 μL固定液重悬细胞,再加入Annexin V和7AAD各2.5 μL,室温避光孵育20 min,应用流式细胞仪检测各组细胞凋亡百分比。
1.7.4 铁死亡标志蛋白检测
将TE1细胞以2×105/孔铺入6孔板,转染体系和时间同上,72 h后提取细胞蛋白,应用Western blotting 检测铁死亡标志蛋白GPX4、FTH1 和ACSL4 表达水平,并以GAPDH为内参照,计算各蛋白表达的相对灰度值。
1.8 统计学处理
采用GraphPad Prism9软件进行统计学分析,采用t检验统计分析食管癌铁死亡抑制基因在TCGA数据和细胞系中的差异,基础实验部分不同分组间的比较采用单因素方差分析,Plt;0.05 为差异有统计学意义。使用Microsoft excel制作统计图。
2 结果
2.1 DEGs筛选
通过R 软件的“limma”包筛选GSE161533 和GSE20347 的DEGs,两个GEO基因数据集分析显示,GSE161533 筛选到4217 个DEGs,其中上调基因1865个,下调基因2352个,GSE20347筛选到3083个DEGs,其中上调基因1463个,下调基因1620个(图1)。
2.2 食管癌与铁死亡相关基因
通过Ferrdb V2数据库共获取铁死亡相关基因484个。将GSE161533、GSE20347数据集获取的DEGs与铁死亡相关基因取交集,共获得58 个食管癌铁死亡基因(图2)。
2.3 GO、KEGG富集和PPI分析
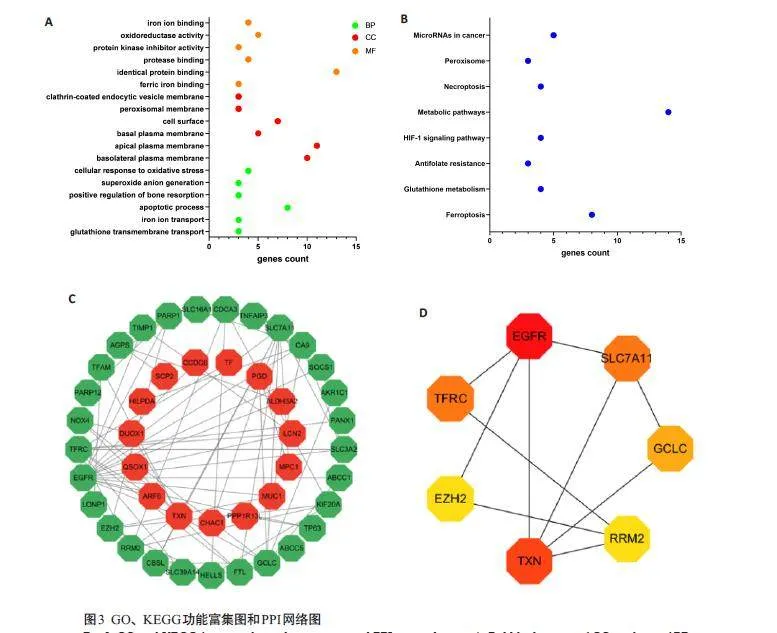
对筛选出的58个食管癌铁死亡基因进行GO分析,发现其在生物学过程方面主要参与谷胱甘肽跨膜转运、铁离子输运、细胞凋亡过程、细胞对氧化应激的反应等,细胞组分方面主要参与基底外侧质膜、顶端质膜和基底质膜形成等,分子功能方面主要参与铁离子结合、蛋白酶结合和氧化还原酶活性等(图3A)。KEGG富集分析显示58 个食管癌铁死亡基因主要参与铁死亡、谷胱甘肽代谢、抗叶酸抵抗、HIF-1信号通路、代谢途径、坏死性凋亡、过氧化物酶体和MicroRNAs与癌症相关信号通路(图3B)。应用STRING数据库构建的PPI网络图包含54个节点和74条边,平均节点连通度为2.55,局部聚类系数为0.522,PPI富集P值小于0.001(图3C)。
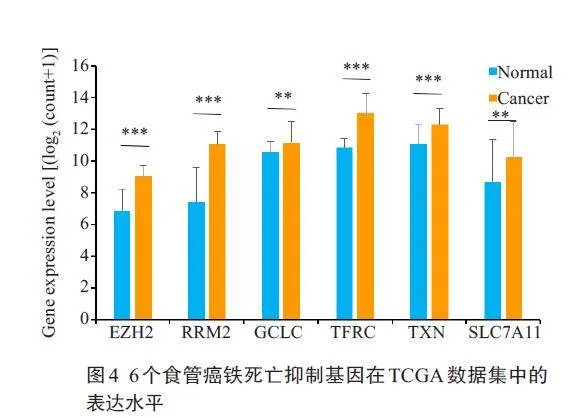
2.4 食管癌铁死亡抑制基因筛选及在TCGA数据集中的表达验证
应用Cytoscape 软件“MCC”、“Degree”和“Closeness”3 种统计学方法共获得7 个核心基因,“Degree”筛选的核心基因网络(图3D)。将核心基因与铁死亡抑制基因取交集后,最终获得6个食管癌铁死亡抑制基因:RRM2、GCLC、TFRC、TXN、SLC7A11、EZH2。
下载TCGA食管癌数据集并筛选,获得食管鳞癌样本95 例,正常食管样本11 例。分析6 个基因在食管鳞癌和正常组织中的表达差异,结果显示:食管鳞癌组织中SLC7A11、GCLC表达水平高于正常食管组织,差异具有统计学意义(Plt;0.01),RRM2、TFRC、TXN、EZH2的表达水平明显高于正常食管组织,差异具有统计学意义(Plt;0.001,图4),与GSE161533 和GSE20347数据集分析的结果一致。
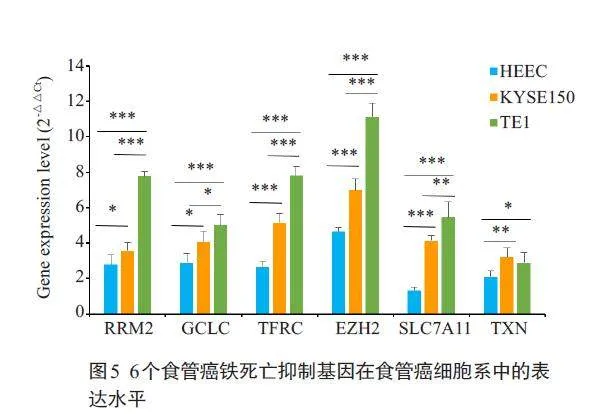
2.5 食管癌铁死亡抑制基因在食管细胞系中的表达
qPCR结果显示:与HEEC细胞比较,食管癌铁死亡抑制基因RRM2、TFRC、TXN、EZH2、SLC7A11 和GCLC mRNA表达水平在TE1细胞和KYSE150细胞中的明显升高,差异有统计学意义(Plt;0.05);与KYSE150细胞比较,RRM2、GCLC、TFRC、SLC7A11 和EZH2mRNA表达水平在TE1细胞中明显升高,差异有统计学意义(Plt;0.05),TXN mRNA表达无变化(图5)。
2.6 下调铁死亡抑制基因使TE1食管癌细胞凋亡增加、增殖能力减弱
流式细胞术和CCK8 实验结果显示:与Control 和Negative组比较,RRM2-siRNA、GCLC-siRNA、TFRCsiRNA、TXN-siRNA、SLC7A11-siRNA和EZH2-siRNA组均可明显促进细胞凋亡,且为晚期凋亡,降低TE1 的细胞增殖能力,差异有统计学意义(Plt;0.05,图6A、B)。
2.7 下调铁死亡抑制基因可诱导TE1 食管癌细胞恢复铁死亡进程
Western blotting 结果显示:与Control 和Negative组比较,RRM2-siRNA、GCLC-siRNA TFRC-siRNA、TXN-siRNA、SLC7A11-siRNA、EZH2-siRNA 组中GPX4、FIH1蛋白表达下调,ACSL4表达上调,差异有统计学意义(Plt;0.05,图6C~F)。
3 讨论
本研究通过对GEO数据库和Ferrdb V2 数据库的筛选,共筛选出58个食管癌铁死亡相关的基因,通过这些基因进行GO分析,我们发现这些基因在多个生物学过程中扮演着重要角色,特别是在GSH跨膜转运、铁离子输运和氧化应激反应等方面。KEGG信号通路分析表明,这些基因参与了铁死亡、GSH代谢和抗叶酸抵抗等关键通路的调节。GSH是一种重要的抗氧化剂,其跨膜转运在细胞膜保护方面具有重要作用,可有效减轻氧化损伤,而肿瘤细胞中GSH过表达可促进肿瘤进展和耐药[17,18] 。一项胃癌的研究表明,生长分化因子15(GDF-15)与多种肿瘤发生有关,GDF-15可以通过上调胃癌细胞的GSH合成诱导化疗耐药性[19] 。铁死亡是一种关键的肿瘤抑制机制,多种肿瘤抑制因子和致癌信号通路已被证明分别促进或抑制铁死亡。p53 是最常见的突变肿瘤抑制因子,在调节铁死亡方面发挥着重要作用[20] 。p53 通过直接结合溶质载体家族7 成员11(SLC7A11)的启动子来降低SLC7A11启动子上的组蛋白H2B单泛素化水平,从而抑制SLC7A11的表达,从而以花生四烯酸12-脂氧合酶 (ALOX12) 依赖的方式使癌细胞对铁死亡敏感[21] 。PIK3CA激活突变或PTEN缺失的癌细胞通常不易受铁死亡的影响[22] 。PI3K 信号通路激活后会促进mTOR 复合物 1 (mTORC1) 的活化,后者可通过促进GPX4蛋白合成、抑制自噬依赖性铁死亡和增加SCD1表达来促进MUFA-PL合成等机制抑制肿瘤细胞铁死亡发生[23,24] 。癌细胞中mTORC1的异常激活促使其免于铁死亡,这或许是PI3K通路突变引发的癌变活性的原因之一。
我们将筛选出的58 个食管癌铁死亡基因导入STRING数据库,分析它们之间的关系并构建PPI网络。结果显示,构建的网络揭示了这些食管癌铁死亡基因之间复杂而多样的关联,凸显了铁死亡调控网络的复杂性和多样性。为进一步从网络中获取关键基因,我们通过Cytoscape的3种统计学方法筛选出了6个食管癌铁死亡抑制基因(RRM2、GCLC、TFRC、TXN、SLC7A11、EZH2),实验验证表明,这些基因在TCGA数据库的食管鳞癌组织和食管癌细胞系中均呈现高表达,且应用siRNA下调这些基因可导致细胞增殖减弱、促进细胞凋亡,恢复细胞的铁死亡进程。因此,这些核心基因可以作为未来食管癌铁死亡机制的候选研究对象,探索它们在食管癌病理生理中的具体作用机制,以及它们是否可以作为潜在的治疗靶点或诊断标志物。
铁死亡在肿瘤治疗中的潜力正逐渐被发掘。许多铁死亡激活剂通过影响铁代谢和利用细胞的铁依赖性来诱导肿瘤细胞死亡。这些激活剂通过多个靶点增加细胞内ROS的产生,提高细胞内氧化应激水平,从而实现抗肿瘤的效果[25] 。索拉菲尼是一种多靶点酪氨酸激酶抑制剂,其主要是通过抑制肿瘤血管生成和抑制RAS/RAF/MEK/ERK信号通路抑制肿瘤生长,广泛应用于肝癌、甲状腺癌等多种肿瘤的治疗[26,27] 。最新研究发现索拉菲尼通过诱导铁死亡抑制肿瘤生长,其通过抑制胱氨酸/谷氨酸反向转运体(System Xc⁻),阻断了胱氨酸的摄取和GSH的生成,从而削弱细胞抗氧化防御能力,由于GSH减少,依赖GSH的GPX4活性下降,无法有效还原脂质过氧化物,导致脂质过氧化物和ROS积累[28] ,高水平的ROS 破坏细胞膜,最终引发细胞膜破裂,导致细胞死亡。Erastin和RSL3是两种经典的铁死亡激活剂,它们通过不同的机制抑制细胞的抗氧化系统。Erastin与索拉菲尼类似,作用于System Xc⁻,阻断胱氨酸的摄取和GSH的生成[29] ,而RSL3 则直接抑制GPX4的活性,二者最终都导致ROS积累和脂质过氧化物的形成[30] 。FIN56和FINO2是另外两种铁死亡激活剂,具有独特的作用机制。FIN56通过诱导GPX4的降解,减少其抗氧化功能[31] 。FINO2不仅直接抑制GPX4的活性,还通过氧化铁增加细胞内的ROS水平,从而加速脂质过氧化,诱导铁死亡[32] 。线粒体是细胞内ROS的重要来源,线粒体功能障碍会导致ROS过量产生,破坏细胞内的抗氧化保护机制平衡,导致氧化应激和DNA损伤,进而促进肿瘤发生和发展。肿瘤细胞通常具有高代谢率和快速增殖的特点,这些特点使得肿瘤细胞更容易产生ROS。因此,合理利用ROS水平的平衡对抑制肿瘤发展是非常重要的。合理的ROS水平能够有效地诱导肿瘤细胞死亡,而过高或过低的ROS水平可能会对肿瘤治疗产生负面影响[33] 。因此,精确调节ROS水平以实现对肿瘤的最佳抑制效果是当前研究的重要方向之一。这些研究不仅揭示了铁死亡在肿瘤治疗中的重要作用,也为开发新型抗肿瘤药物提供了理论基础。未来的研究可以进一步探索这些激活剂的具体机制,寻找新的靶点和组合疗法,以提高抗肿瘤治疗效果。
综上所述,铁死亡机制在食管癌中研究较少,本文也仅在细胞学实验中证实了基因表达变化对食管癌细胞功能和铁死亡功能恢复的影响,后期可在此基础上,继续探索这些基因在食管癌铁死亡中的调控机制,以期为食管癌的发病机制和后续靶向治疗提供新的研究方向。
参考文献:
[1] Short MW, Burgers KG, Fry VT. Esophageal cancer[J]. Am FamPhysician, 2017, 95(1): 22-8.
[2] Liu Y, Ge QQ, Xu SN, et al. Efficacy and safety of anlotinib plusprogrammed death-1 blockade versus anlotinib monotherapy assecond or further-line treatment in advanced esophageal squamouscell carcinoma: a retrospective study[J]. Front Oncol, 2022, 12:942678.
[3] Hong T, Lei G, Chen X, et al. PARP inhibition promotes ferroptosisvia repressing SLC7A11 and synergizes with ferroptosis inducers inBRCA-proficient ovarian cancer[J]. Redox Biol, 2021, 42: 101928.
[4] Cheng Q, Bao LJ, Li MQ, et al. Erastin synergizes with cisplatin viaferroptosis to inhibit ovarian cancer growth in vitro and in vivo[J]. JObstet Gynaecol Res, 2021, 47(7): 2481-91.
[5] Yin LZ, Liu PY, Jin Y, et al. Ferroptosis-related small-moleculecompounds in cancer therapy: strategies and applications[J]. Eur JMed Chem, 2022, 244: 114861.
[6] Guo JP, Xu BF, Han Q, et al. Ferroptosis: a novel anti-tumor actionfor cisplatin[J]. Cancer Res Treat, 2018, 50(2): 445-60.
[7] Tao H, Song SJ, Fan ZW, et al. PKCiota inhibits the ferroptosis ofesophageal cancer cells via suppressing USP14-mediatedautophagic degradation of GPX4[J]. Antioxidants, 2024, 13(1): 114.
[8] Fan X, Fan YT, Zeng H, et al. Role of ferroptosis in esophagealcancer and corresponding immunotherapy[J]. World J GastrointestOncol, 2023, 15(7): 1105-18.
[9] Shishido Y, Amisaki M, Matsumi Y, et al. Antitumor effect of 5-aminolevulinic acid through ferroptosis in esophageal squamous cellcarcinoma[J]. Ann Surg Oncol, 2021, 28(7): 3996-4006.
[10] Jiang B, Zhao YQ, Shi M, et al. DNAJB6 promotes ferroptosis inesophageal squamous cell carcinoma[J]. Dig Dis Sci, 2020, 65(7):1999-2008.
[11]Maimaitizunong R, Wang K, Li H. Ferroptosis and its emerging rolein esophageal cancer[J]. Front Mol Biosci, 2022, 9: 1027912.
[12]Yao WJ, Wang JJ, Meng FR, et al. Circular RNA CircPVT1 inhibits5-fluorouracil chemosensitivity by regulating ferroptosis throughmiR-30a-5p/FZD3 axis in esophageal cancer cells[J]. Front Oncol,2021, 11: 780938.
[13]Zhang JH, Wang N, Zhou YY, et al. Oridonin induces ferroptosis byinhibiting gamma-glutamyl cycle in TE1 cells[J]. Phytother Res,2021, 35(1): 494-503.
[14]Yang RY, Chen FZ, Xu HZ, et al. Exploring the mechanism ofrealgar against esophageal cancer based on ferroptosis induced byROS-ASK1-p38 MAPK signaling pathway[J]. Evid BasedComplement Alternat Med, 2022: 3698772.
[15]Yin LB, Li ZW, Wang JL, et al. Sulfasalazine inhibits esophagealcancer cell proliferation by mediating ferroptosis[J]. Chem BiolDrug Des, 2023, 102(4): 730-7.
[16]Xia Y, Tang YX, Huang ZX, et al. Artesunate-loaded solid lipidnanoparticles resist esophageal squamous cell carcinoma byinducing Ferroptosis through inhibiting the AKT/mTOR signaling[J]. Cell Signal, 2024, 117: 111108.
[17]Niu BY, Liao KX, Zhou YX, et al. Application of glutathionedepletion in cancer therapy: enhanced ROS-based therapy,ferroptosis, and chemotherapy[J]. Biomaterials, 2021, 277: 121110.
[18]Criscuolo D, Avolio R, Parri M, et al. Decreased levels of GSH areassociated with platinum resistance in high-grade serous ovariancancer[J]. Antioxidants, 2022, 11(8): 1544.
[19]Wang SF, Chang YL, Fang WL, et al. Growth differentiation factor15 induces cisplatin resistance through upregulation of xCTexpression and glutathione synthesis in gastric cancer[J]. CancerSci, 2023, 114(8): 3301-17.
[20]Xu R, Wang WN, Zhang WL. Ferroptosis and the bidirectionalregulatory factor p53[J]. Cell Death Discov, 2023, 9(1): 197.
[21]Chu B, Kon N, Chen DL, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway[J]. Nat Cell Biol, 2019, 21(5): 579-91.
[22]Yi JM, Zhu JJ, Wu J, et al. Oncogenic activation of PI3K-AKTmTORsignaling suppresses ferroptosis via SREBP-mediatedlipogenesis[J]. Proc Natl Acad Sci USA, 2020, 117(49): 31189-97.
[23]Lei G, Zhuang L, Gan BY. mTORC1 and ferroptosis: regulatorymechanisms and therapeutic potential[J]. Bioessays, 2021, 43(8):e2100093.
[24]Zhang YL, Swanda RV, Nie LT, et al. mTORC1 couples cyst(e)ineavailability with GPX4 protein synthesis and ferroptosis regulation[J]. Nat Commun, 2021, 12(1): 1589.
[25]Ma SM, Dielschneider RF, Henson ES, et al. Ferroptosis andautophagy induced cell death occur independently after siramesineand lapatinib treatment in breast cancer cells[J]. PLoS One, 2017, 12(8): e0182921.
[26]Man SL, Luo C, Yan MY, et al. Treatment for liver cancer: fromsorafenib to natural products[J]. Eur J Med Chem, 2021, 224:113690.
[27]Fallahi P, Ferrari SM, Galdiero MR, et al. Molecular targets oftyrosine kinase inhibitors in thyroid cancer[J]. Semin Cancer Biol,2022, 79: 180-96.
[28]Li QH, Chen KX, Zhang TY, et al. Understanding sorafenib-inducedferroptosis and resistance mechanisms: implications for cancertherapy[J]. Eur J Pharmacol, 2023, 955: 175913.
[29]Costa I, Barbosa DJ, Benfeito S, et al. Molecular mechanisms offerroptosis and their involvement in brain diseases[J]. PharmacolTher, 2023, 244: 108373.
[30]Sui XB, Zhang RN, Liu SP, et al. RSL3 drives ferroptosis throughGPX4 inactivation and ROS production in colorectal cancer[J].Front Pharmacol, 2018, 9: 1371.
[31]Sun YD, Berleth N, Wu WX, et al. Fin56-induced ferroptosis issupported by autophagy-mediated GPX4 degradation and functionssynergistically with mTOR inhibition to kill bladder cancer cells[J].Cell Death Dis, 2021, 12(11): 1028.
[32]Gaschler MM, Andia AA, Liu HR, et al. FINO2 initiates ferroptosisthrough GPX4 inactivation and iron oxidation[J]. Nat Chem Biol,2018, 14(5): 507-15.
[33]Cheung EC, Vousden KH. The role of ROS in tumour developmentand progression[J]. Nat Rev Cancer, 2022, 22(5): 280-97.
(编辑:吴锦雅)
基金项目:天津市自然科学基金京津冀基础研究合作专项(20JCZXJC00190)
猜你喜欢
医学信息(2016年36期)2017-02-23 14:00:06
中国中药杂志(2016年22期)2017-02-13 17:28:52
中国当代医药(2016年19期)2016-09-30 20:43:59
科技视界(2016年15期)2016-06-30 12:27:37
中国实用医药(2016年11期)2016-05-04 21:56:32
成长·读写月刊(2015年9期)2015-11-09 21:32:14
中国医药导报(2015年26期)2015-10-16 20:43:00
山东体育学院学报(2015年3期)2015-08-14 20:30:25
中国当代医药(2015年18期)2015-08-06 18:09:34
中国现代医生(2015年13期)2015-06-17 10:19:05
