
丁酸钠与索拉非尼可能通过YAP 诱导铁死亡协同抑制肝癌细胞增殖
2024-11-03 00:00:00何华星刘璐琳刘颖茵陈纳川孙素霞
南方医科大学学报 2024年7期


摘要:目的 探讨丁酸钠(NaB)联合索拉非尼(Sora)是否协同诱导铁死亡抑制肝癌细胞增殖及其机制。方法 CCK8、平板克隆实验和倒置光学显微镜观察探讨NaB或(和)Sora 对HepG2 肝癌细胞增殖活性的影响;GSH法和C11-BODIPY 581/591 检测NaB或(和)Sora是否诱导HepG2细胞发生铁死亡;公共数据库TCGA分析肝癌组织与正常组织中YAP基因表达差异;Westernblotting检测NaB或(和)Sora对HepG2细胞中YAP蛋白表达及其磷酸化水平的影响。结果 CCK8结果显示,2 mmol/L NaB联合Sora可显著降低HepG2肝癌细胞对Sora的IC50 (Plt;0.001),联合指数法证实Sora与NaB联合具有协同作用(CIlt;1),铁死亡抑制剂(Fer-1)和YAP激活剂(XMU)则可逆转NaB联合Sora对HepG2细胞的增殖抑制作用(Plt;0.001、Plt;0.001);平板克隆实验结果显示,相较NaB或Sora单独处理,NaB联合Sora可显著抑制HepG2细胞克隆球形成(Plt;0.01,Plt;0.01);形态学观察结果显示,NaB或Sora单独处理均可使HepG2细胞分散,形态皱缩且数量减少,而两者联合处理可加剧这一现象;GSH法和C11-BODIPY581/591检测结果显示,较NaB或Sora单独处理,NaB联合Sora可进一步降低细胞内GSH水平(Plt;0.001,Plt;0.05)和脂质ROS水平(Plt;0.05,Plt;0.01),Fer-1和XMU可逆转NaB联合Sora对HepG2细胞内GSH水平的下调作用(Plt;0.001,Plt;0.01)和脂质ROS水平的上调作用(Plt;0.01,Plt;0.05);TCGA分析结果显示,肝癌组织与正常组织相比YAP mRNA高表达(Plt;0.001);Westernblotting 结果显示,较NaB或Sora 单独处理,NaB联合Sora 进一步下调HepG2 细胞YAP蛋白表达水平(Plt;0.05,Plt;0.05),上调YAP蛋白磷酸化水平(Plt;0.05,Plt;0.01)。结论 NaB联合Sora可能通过抑制YAP诱导铁死亡,进而协同抑制肝癌细胞增殖。
关键词:丁酸钠;索拉非尼;肝癌;铁死亡;YAP;细胞增殖
全球有超过900万人被诊断为原发性肝癌,是6大最常诊断的癌症之一,也是癌症相关死亡的第4 大原因[1],严重危害我国乃至世界人民的生命健康[2]。索拉非尼(Sora)是第1 个用于晚期肝癌一线治疗的靶向药物,但由于其耐药性、副作用大和总体疗效有限[3],临床上迫切需要寻找有效的联合疗法以改善其治疗效果。丁酸钠(NaB)是膳食纤维在肠道菌群作用下分解产生的一种短链脂肪酸,具有广泛的生物活性,现已被用于研究治疗多种疾病如炎症性肠病、非酒精性脂肪肝[4]。NaB作为一种组蛋白去乙酰化酶抑制剂,可以促进抗癌基因表达,从而抑制肿瘤细胞增殖[5, 6],还能通过调控铁死亡相关基因来发挥抗癌效果[7]。
铁死亡是一种铁依赖性的新型细胞死亡方式,不同于细胞凋亡、自噬和坏死,其特征为还原型谷胱甘肽(GSH)的减少和脂质ROS的大量积累[8]。Sora 的抗癌活性依赖于通过抑制谷氨酸/胱氨酸逆转运蛋白(SLC7A11)的活性来诱导铁死亡[9]。课题组前期研究表明,NaB也可通过诱导铁死亡的方式抑制结直肠癌和肝癌细胞的增殖[7, 10]。由于Sora 和NaB都能靶向肝癌细胞铁死亡,我们推测两者可能通过协同诱导铁死亡来抑制肝癌细胞增殖。然而,目前关于Sora 与NaB联合用药的报道极少,Yu等[11]发现NaB可减轻LM3肝癌细胞有氧糖酵解,进而增强Sora 的敏感性。但有关Sora联合NaB是否协同抑制HepG2肝癌细胞增殖以及是否协同诱导铁死亡尚未有研究。此外,有研究报道,促进Yes 相关蛋白(YAP)介导的铁死亡可提高肝癌对Sora敏感性,这进一步揭示了本研究的意义[12]。
YAP是Hippo信号通路的最终效应分子,参与肿瘤发生和组织再生等多种生理病理过程[13]。近年来的研究表明,YAP蛋白的高表达是各种癌症化疗产生耐药性的重要因素[14,15]。同时,YAP及其磷酸化水平通过调节参与维持细胞内ROS和脂质过氧化平衡的基因表达来调节铁死亡[16]。目前,有关YAP在Sora 联合NaB抑制肝癌细胞中的作用尚未见报道。本研究旨在研究Sora联合NaB是否通过协同诱导铁死亡抑制HepG2肝癌细胞增殖,并初步探讨YAP对两者联合诱导铁死亡的影响及机制,为肝癌的治疗提供新的思路。
1 材料和方法
1.1 细胞与试剂
人肝癌细胞系HepG2 细胞(中科院上海生物细胞库);NaB、二甲基亚砜(DMSO)、结晶紫(Sigma);Sora、GSH检测试剂盒(北京索莱宝);DMEM培养基、胰酶(Gibco)、胎牛血清(大连美仑);CCK8(Targetmol);铁死亡抑制剂(Fer-1)(Glpbio);YAP 激活剂(XMU,上海陶术);GAPDH、羊抗兔、羊抗鼠(北京锐抗);C11-BODIPY 581/591(CAYMAN CHEMICAL)。
1.2 细胞培养
使用含10%胎牛血清的DMEM培养基,于37 ℃、5%CO2饱和湿度培养箱中培养,取对数生长期细胞进行后续细胞实验。
1.3 CCK8法
将对数生长期的细胞以5000/孔的密度接种至96孔板,37 ℃、5%CO2饱和湿度培养箱中过夜培养,待细胞贴壁后,加入不同浓度的(0.5、1、2、4、8、16 μmol/L)Sora或(和)2 mmol/L NaB处理24 h。随后弃去培养基,加入10 μL CCK8 溶液和100 μL完全培养基,孵育1h后在450 nm波长处测定吸光度,计算细胞活力。细胞活力(%)=[A450(样品)-A450(空白)]/[A450(对照)-A450(空白)]×100%。
1.4 平板克隆实验
对数生长期的HepG2 细胞在NaB或(和)Sora 或(和)Fer-1处理24 h后,胰酶消化并重悬。以2000/孔的密度接种于新的6 孔板,隔2~4 d 换液,待绝大多数单个克隆的细胞数超过50个时终止培养,弃培养基,并用PBS溶液清洗2 次。每孔加入1 mL 4%多聚甲醛固定30 min,再用PBS清洗2次。最后,用0.1%结晶紫染色15 min,流水清洗,自然晾干后拍照,并通过ImageJ软件进行克隆球计数。
1.5 GSH检测
对数生长期的HepG2 细胞在NaB或(和)Sora 或(和)Fer-1处理24 h后,胰酶消化后用PBS清洗2次,收集细胞并按GSH试剂盒说明,用3倍体积的试剂一重悬混匀,冻融3 次后以8000 g/min 离心10 min,收集上清液用于后续测定。在412 nm波长处用酶标仪测定空白样及不同浓度标准样品的吸光度,绘制标准曲线并测定各组样品的GSH浓度。
1.6 脂质ROS检测
对数生长期的HepG2 细胞在NaB或(和)Sora 或(和)Fer-1处理24 h后,弃培养基,用HBSS清洗2次,加入无血清DMEM培养基和C11-BODIPY 581/591工作液(按说明书配制),置避光孵育30 min后弃上清,再次用HBSS清洗2 次后加入HBSS,用荧光显微镜观察红色(还原型)和绿色(氧化型)荧光强度。还原型:Ex=561 nm,Em=600~630 nm;氧化型:Ex=488 nm,Em=510~550 nm。使用Image J软件进行荧光定量,相对平均荧光强度=该区域绿色荧光强度总和/该区域红色荧光强度总和。
1.7 Western blotting实验
处理后的各组HepG2 细胞用预冷PBS 清洗2 次,加入适量RIPA裂解液,冰上静置15 min后离心收集蛋白并定量,加入5×buffer 煮沸10 min。配制10% 的SDS-PAGE凝胶。样品上样、电泳、转膜,5%牛奶封闭2 h后孵育一抗,4 ℃过夜。TBST洗涤3次后加入二抗,室温孵育2 h,再次用TBST洗涤3次后用ECL化学发光法显影。通过ImageJ软件分析条带灰度值。
1.8 统计学方法
数据分析使用SPSS 26.0 和Graph Pad Prism 8.0。符合正态分布的计量资料以均数±标准差表示,两组均数比较采用t检验,多组均数比较采用单因素方差分析,Plt;0.05为差异有统计学意义。在中位剂量效应分析中使用Compusyn软件计算联合指数(CI)。
2 结 果
2.1 NaB增强Sora对HepG2肝癌细胞增殖的抑制作用
CCK8 结果显示,1 μmol/L Sora 与2 mmol/L NaB联合处理24 h 可显著抑制HepG2 细胞增殖(Plt;0.001,图1A)。相比单独使用Sora,联合处理将Sora的IC50从12.88±0.90 μmol/L 显著降低至2.83±0.10 μmol/L(Plt;0.001,图1A)。中位剂量效应分析表明,联合处理的CI值均小于1(图1A),NaB与Sora联合具有协同作用。在后续实验中,选用2 mmol/L NaB和4 μmol/L Sora,联合处理的抑制率达(52.84±3.59)%(图1A),CI值低至0.58(图1A)。平板克隆实验结果显示,与对照组相比,NaB或Sora单独处理均可抑制HepG2细胞的克隆形成能力(Plt;0.01,Plt;0.05,图1B),联合处理进一步显著抑制HepG2 细胞的克隆形成能力(Plt;0.01,Plt;0.01,图1B)。倒置光学显微镜观察显示,与对照组相比,单独处理的细胞呈分散,皱缩形态且数量减少,而联合处理加剧了这些现象(图1 C)。
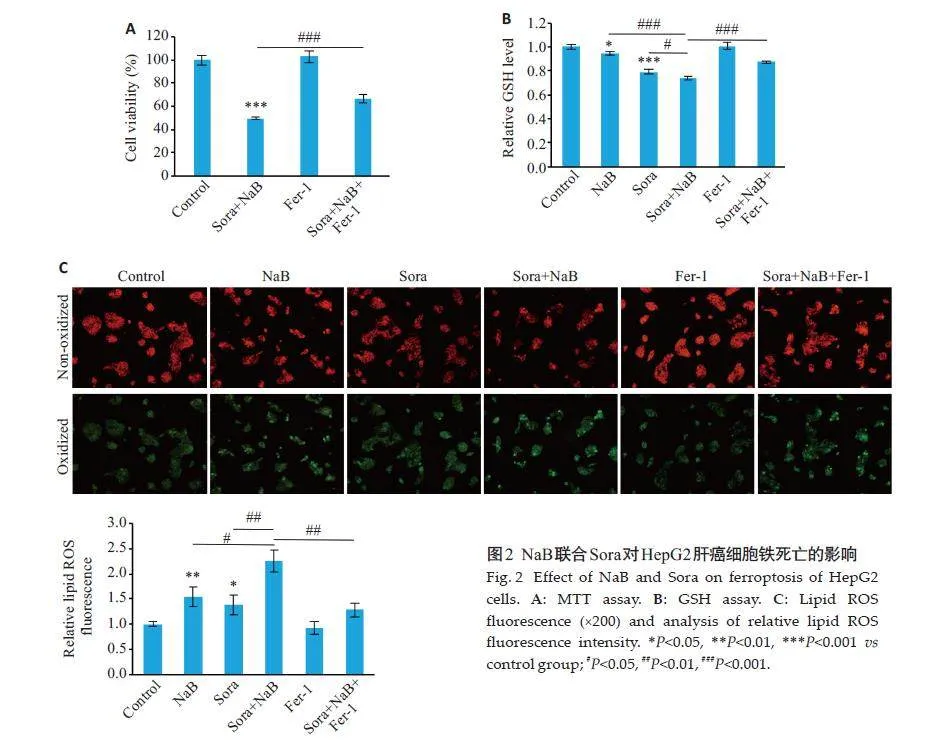
2.2 NaB联合Sora对HepG2肝癌细胞铁死亡的影响
CCK8 结果显示,铁死亡抑制剂Fer-1 可逆转NaB联合Sora 对HepG2 细胞的增殖抑制作用(Plt;0.001,图2A)。GSH水平检测结果显示,与对照组相比,NaB或Sora单独处理可降低HepG2细胞的GSH水平(Plt;0.05,Plt;0.001,图2B),联合处理则进一步降低GSH 水平(Plt;0.001,Plt;0.05,图2B),且此降低效应可被Fer-1恢复(Plt;0.001,图2B)。脂质ROS荧光结果显示,与对照组相比,NaB或Sora 单独处理可增加HepG2 细胞的脂质ROS水平(Plt;0.01,Plt;0.05,图2C),而较单独NaB或Sora 组,联合处理组进一步显著增加细胞内脂质ROS 水平(Plt;0.05,Plt;0.01,图2C),且此效应也可被Fer-1恢复(Plt;0.01,图2C)。
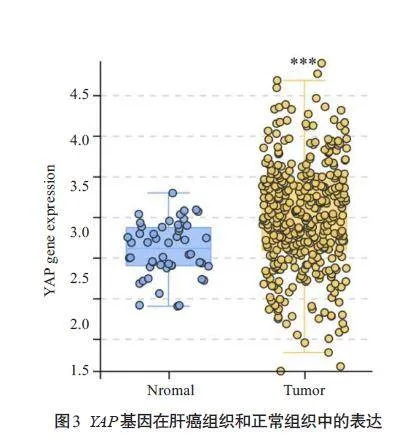
2.3 YAP在肝癌组织中高表达
TCGA公共数据库分析结果显示,与正常肝脏组织相比,肝癌组织中YAP mRNA 水平表达更高(Plt;0.001,图3)。
2.4 NaB 联合Sora 对HepG2 肝癌细胞内的YAP 和p-YAP表达的影响
Western blotting结果显示,与对照组相比,NaB可下调HepG2细胞中的YAP蛋白表达水平(Plt;0.01,图4A、B),并上调p-YAP蛋白表达水平(Plt;0.05,图4A、B)。同样,Sora下调YAP蛋白表达水平(Plt;0.05,图4A、B),并上调p-YAP蛋白表达水平(Plt;0.05,图4A、B)。相比单独NaB或Sora处理,联合处理显著降低YAP蛋白表达水平(Plt;0.05,Plt;0.05,图4A、B),同时显著增加p-YAP蛋白水平(Plt;0.05,Plt;0.01,图4A、B)。
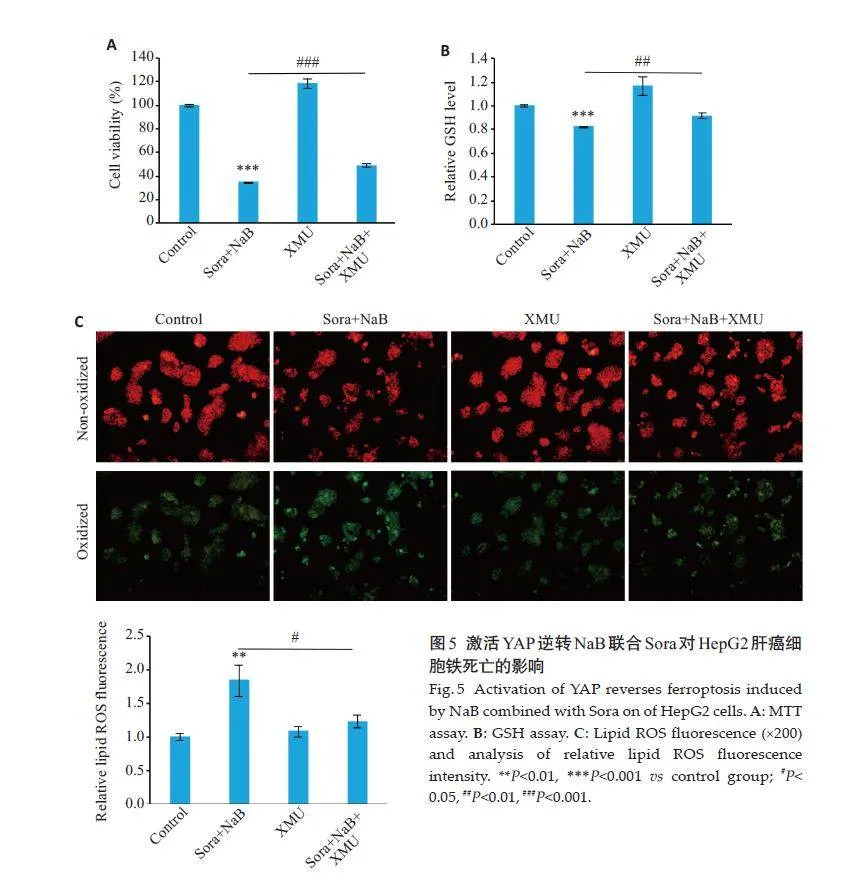
2.5 激活YAP逆转NaB联合Sora对HepG2肝癌细胞增殖和铁死亡的影响
CCK8 结果显示,预处理3 μmol/L XMU后,NaB联合Sora 对HepG2 细胞的增殖抑制作用被部分逆转(Plt;0.001,图5A)。GSH检测结果显示,XMU可恢复联合处理引起的GSH水平下降(Plt;0.01,图5B)。脂质ROS荧光结果显示,与联合处理组相比,XMU预处理后联合处理组HepG2细胞的平均荧光强度显著降低(Plt;0.05,图5C)。
3 讨 论
近年来,生物活性物质联合化疗药物在肝癌治疗中的应用受到广泛关注。例如,槲皮素、白藜芦醇和姜黄素与Sora的联用均表现出显著增强的抗肝癌作用,能够有效抑制肿瘤细胞增殖并诱导凋亡[17-19]。在本研究中,NaB与Sora 的联合应用展示了类似的协同效应,能更有效地抑制HepG2 肝癌细胞的增殖。不同的是,我们还研究了两者联合对铁死亡的影响。铁死亡作为治疗耐药性肿瘤的潜在靶点,在NaB与Sora 协同效应中的作用尚未被研究。
铁死亡的经典通路中,SLC7A11 介导胱氨酸的摄取和谷氨酸的释放减少,从而导致GSH合成减少[20]。GSH作为重要的细胞内抗氧化剂,维持细胞的氧化还原稳态,其减少会破坏这种稳态并导致脂质ROS 积累[21],最终诱导细胞的铁死亡。我们的研究结果显示,相较于单药处理,Sora 联合NaB可显著降低了HepG2细胞的GSH水平,并增加脂质ROS含量。在加入Fer-1后,这些效应得以逆转,同时联合处理导致的增殖抑制作用也被逆转。这些结果表明,Sora 联合NaB可通过协同诱导铁死亡的方式抑制HepG2细胞增殖。
在抵抗铁死亡过程中,YAP 蛋白发挥了重要作用[22]。与对Sora敏感的细胞相比,Sora耐药细胞中YAP表达水平更高,且对Sora诱导的铁死亡耐受性更强[12]。阻断YAP表达不仅可以有效诱导细胞的氧化损伤[23],还可以使肝癌细胞重新获得对铁死亡的敏感性[24]。在本研究中,Sora 联合NaB可显著降低YAP蛋白的表达水平,并促进YAP磷酸化。正常情况下,磷酸化的YAP会在细胞质中被降解,而未被磷酸化的YAP会进入细胞核中发挥其转录调控活性,促进其下游的铁死亡负性调控分子如SLC7A11的基因表达,进而降低肝癌细胞铁死亡敏感性[12, 24]。我们推测Sora联合NaB可能通过共同降低YAP表达、促进其磷酸化来诱导肝癌细胞的铁死亡。然而,也有研究认为YAP具有相反的作用[25, 26]。
为进一步探索YAP在Sora联合NaB诱导肝癌细胞铁死亡中的机制,我们引入了YAP激活剂XMU。XMU选择性抑制YAP上游信号分子蛋白激酶Mst1/2,提高YAP的表达,抑制YAP磷酸化,从而增强其在细胞核内的转录调控作用[27, 28]。后续实验结果显示,XMU的加入有效逆转了Sora联合NaB对HepG2细胞铁死亡的影响。这些研究及实验结果提示,NaB和Sora 可能通过抑制YAP表达、促进其磷酸化来协同诱导肝癌细胞的铁死亡,进而抑制肝癌细胞增殖。
综上所述,NaB联合Sora可能通过调节YAP介导的铁死亡途径,从而协同抑制肝癌细胞的增殖,为开发更为有效的肝癌靶向治疗提供新的思路和依据。
参考文献:
[1] Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020:GLOBOCAN estimates of incidence and mortality worldwide for 36cancers in 185 countries[J]. CA Cancer J Clin, 2021, 71(3): 209-49.
[2] Donne R, Lujambio A. The liver cancer immune microenvironment:therapeutic implications for hepatocellular carcinoma[J].Hepatology, 2023, 77(5): 1773-96.
[3] Villanueva A. Hepatocellular carcinoma[J]. N Engl J Med, 2019,380(15): 1450-62.
[4] Kaźmierczak-Siedlecka K, Marano L, Merola E, et al. Sodiumbutyrate in both prevention and supportive treatment of colorectalcancer[J]. Front Cell Infect Microbiol, 2022, 12: 1023806.
[5] Bultman SJ. Molecular pathways: gene-environment interactionsregulating dietary fiber induction of proliferation and apoptosis viabutyrate for cancer prevention[J]. Clin Cancer Res, 2014, 20(4):799-803.
[6] D'Argenio G, Cosenza V, Delle Cave M, et al. Butyrate enemas inexperimental colitis and protection against large bowel cancer in arat model[J]. Gastroenterology, 1996, 110(6): 1727-34.
[7] Bian ZB, Sun XD, Liu LL, et al. Sodium butyrate induces CRC cellferroptosis via the CD44/SLC7A11 pathway and exhibits a synergistictherapeutic effect with erastin[J]. Cancers, 2023, 15(2): 423.
[8] Lei G, Zhuang L, Gan BY. Targeting ferroptosis as a vulnerability incancer[J]. Nat Rev Cancer, 2022, 22: 381-96.
[9] Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition ofcystine-glutamate exchange induces endoplasmic reticulum stressand ferroptosis[J]. Elife, 2014, 3: e02523.
[10]孙小蝶, 秦 勇, 刘璐琳, 等. 丁酸钠通过诱导铁死亡抑制肝癌细胞增殖[J]. 营养学报, 2023, 45(2): 157-62.
[11]Yu Q, Dai WQ, Ji J, et al. Sodium butyrate inhibits aerobicglycolysis of hepatocellular carcinoma cells via the c-myc/hexokinase 2 pathway[J]. J Cell Mol Med, 2022, 26(10): 3031-45.
[12]Gao RZ, Kalathur RKR, Coto-Llerena M, et al. YAP/TAZ and ATF4drive resistance to Sorafenib in hepatocellular carcinoma bypreventing ferroptosis[J]. EMBO Mol Med, 2021, 13(12): e14351.
[13]Franklin JM, Wu ZM, Guan KL. Insights into recent findings andclinical application of YAP and TAZ in cancer[J]. Nat Rev Cancer,2023, 23: 512-25.
[14]Marti P, Stein C, Blumer T, et al. YAP promotes proliferation,chemoresistance, and angiogenesis in human cholangiocarcinomathrough TEAD transcription factors[J]. Hepatology, 2015, 62(5):1497-510.
[15]Mao B, Hu F, Cheng J, et al. SIRT1 regulates YAP2-mediated cellproliferation and chemoresistance in hepatocellular carcinoma[J].Oncogene, 2014, 33(11): 1468-74.
[16]Zhou W, Lim A, Edderkaoui M, et al. Role of YAP signaling inregulation of programmed cell death and drug resistance in cancer[J]. Int J Biol Sci, 2024, 20(1): 15-28.
[17]Zhang ZG, Wu HT, Zhang YJ, et al. Dietary antioxidant quercetinovercomes the acquired resistance of Sorafenib in Sorafenibresistanthepatocellular carcinoma cells through epidermal growthfactor receptor signaling inactivation[J]. Naunyn SchmiedebergsArch Pharmacol, 2024, 397(1): 559-74.
[18]Gao ML, Deng C, Dang F. Synergistic antitumor effect of resveratroland sorafenib on hepatocellular carcinoma through PKA/AMPK/eEF2K pathway[J]. Food Nutr Res, 2021, 65: 1-14.
[19]Cao HQ, Wang YX, He XY, et al. Codelivery of sorafenib andcurcumin by directed self-assembled nanoparticles enhancestherapeutic effect on hepatocellular carcinoma[J]. Mol Pharm,2015, 12(3): 922-31.
[20]Chen X, Li JB, Kang R, et al. Ferroptosis: machinery and regulation[J]. Autophagy, 2021, 17(9): 2054-81.
[21]Niu BY, Liao KX, Zhou YX, et al. Application of glutathionedepletion in cancer therapy: enhanced ROS-based therapy,ferroptosis, and chemotherapy[J]. Biomaterials, 2021, 277: 121110.
[22]Sun T, Chi JT. Regulation of ferroptosis in cancer cells by YAP/TAZand Hippo pathways: the therapeutic implications[J]. Genes Dis,2021, 8(3): 241-9.
[23]Yu HF, Yang ZQ, Xu MY, et al. Yap is essential for uterinedecidualization through Rrm2/GSH/ROS pathway in response toBmp2[J]. Int J Biol Sci, 2022, 18(6): 2261-76.
[24]Chen JS, Zhang J, Tian W, et al. AKR1C3 suppresses ferroptosis inhepatocellular carcinoma through regulation of YAP/SLC7A11signaling pathway[J]. Mol Carcinog, 2023, 62(6): 833-44.
[25]Qin YF, Pei Z, Feng Z, et al. Oncogenic activation of YAP signalingsensitizes ferroptosis of hepatocellular carcinoma via ALOXE3-mediated lipid peroxidation accumulation[J]. Front Cell Dev Biol,2021, 9: 751593.
[26]Wu J, Minikes AM, Gao MH, et al. Intercellular interaction dictatescancer cell ferroptosis via NF2-YAP signalling[J]. Nature, 2019,572(7769): 402-6.
[27]Chen TQ, Sun DD, Wang QJ, et al. α‑hederin inhibits theproliferation of hepatocellular carcinoma cells via hippo-yes-associatedprotein signaling pathway[J]. Front Oncol, 2022, 12: 839603.
[28]Fan FQ, He ZX, Kong LL, et al. Pharmacological targeting ofkinases MST1 and MST2 augments tissue repair and regeneration[J]. Sci Transl Med, 2016, 8(352): 352ra108.
(编辑:余诗诗)
基金项目:国家自然科学基金(81773429);广东省自然科学基金(2022A1515011631,2024A1515012175)
猜你喜欢
天津医科大学学报(2021年1期)2021-01-26 00:57:10
天津医科大学学报(2019年3期)2019-08-13 06:53:08
右江医学(2016年4期)2017-01-05 16:26:48
医学信息(2016年29期)2016-11-28 09:54:46
课程教育研究·学法教法研究(2016年6期)2016-04-26 10:06:57
中国继续医学教育(2015年3期)2016-01-06 01:36:44
分子影像学杂志(2015年3期)2015-12-04 03:28:58
现代养生·下半月(2015年8期)2015-11-16 20:03:43
肿瘤预防与治疗(2015年1期)2015-09-26 07:26:20
中国当代医药(2015年16期)2015-03-01 02:03:11
