
混合性结缔组织病相关肺动脉高压临床特点分析
2024-07-13 07:18王慧潘晴王宙明张娜杨振文魏蔚
天津医药 2024年7期
王慧 潘晴 王宙明 张娜 杨振文 魏蔚

作者单位:1天津医科大学总医院风湿免疫科(邮编300052),2心血管内科
作者简介:王慧(1990),女,主治医师,主要从事结缔组织病相关肺损害方面研究。E-mail:tjwhdyx@163.com
△通信作者 E-mail:tjweiwei2003@163.com
摘要:目的 探究混合性结缔组织病相关肺动脉高压(MCTD-PAH)患者的临床特点及发病危险因素。方法 回顾性纳入12例住院治疗的MCTD-PAH患者(MCTD-PAH组),根据性别、年龄按1︰3随机抽取同期住院的36例混合性结缔组织病无肺动脉高压(MCTD-non-PAH)患者作为对照组,比较2组患者的临床表现和辅助检查,随诊2组患者生存状态。结果 MCTD-PAH组较对照组出现活动后气短、肌炎及心包积液比例更高,血沉及免疫球蛋白G(IgG)水平更高。多因素Logistic回归分析显示,活动后气短及较高水平的IgG是预测MCTD发生PAH的危险因素。MCTD-PAH死亡3例(16.7%),对照组无患者死亡。结论 PAH是MCTD严重的并发症之一,MCTD患者出现活动后气短及较高水平的IgG时需警惕合并PAH。
关键词:混合性结缔组织病;肺动脉高压;右心导管;临床特点
中图分类号:R544.16,R593.27文献标志码:ADOI:10.11958/20231786
Analysis of clinical features of mixed connective tissue disease associated with pulmonary arterial hypertension
WANG Hui1, PAN Qing1, WANG Zhouming2, ZHANG Na1, YANG Zhenwen2, WEI Wei1△
1 Department of Rheumatology and Immunology, 2 Department of Cardiovasology, Tianjin Medical University
General Hospital, Tianjin 300052, China
△Corresponding Author E-mail: tjweiwei2003@163.com
Abstract: Objective To investigate the clinical characteristics and risk factors of mixed connective tissue disease associated with pulmonary arterial hypertension (MCTD-PAH). Methods Twelve MCTD-PAH patients diagnosed by right heart catheterization (RHC) at Tianjin Medical University General Hospital were retrospectively included, and 36 MCTD patients without pulmonary arterial hypertension (MCTD-non-PAH) were randomly selected from the same period of hospitalization based on gender and age. The clinical features and auxiliary examination of the two groups were compared, and the survival status of the two groups was compared. Results The proportion of dyspnea after activity, myositis and pericardial effusion were higher in the MCTD-PAH group than those of the control group. Serum sedimentation rate and immunoglobulin G (IgG) levels were higher in the MCTD-PAH group. Multivariate Logistic regression analysis showed that dyspnea after activity and high level of IgG were risk factors for predicting the occurrence of PAH in MCTD. Three patients (16.7%) died in the MCTD-PAH group, and no patients died in the control group. Conclusion Pulmonary arterial hypertension is one of the serious complications of MCTD. MCTD patients have shortness of breath after activity and high level of IgG should be wary of concomitant PAH.
Key words: mixed connective tissue disease; pulmonary arterial hypertension; right heart catheterization; clinical features
混合性结缔组织病(mixed connective tissue disease,MCTD)是一种慢性自身免疫病,常有手肿胀、滑膜炎、肌炎、雷诺现象、肢端硬化等一种或多种临床表现,以血清高滴度斑点型抗核抗体(ANA)和高滴度抗U1核糖核蛋白(U1RNP)抗体阳性、抗Sm抗体阴性为特点。肺损害是MCTD患者常见的器官损害之一,也是影响预后的重要因素,以间质性肺病(interstitial lung disease,ILD)及肺动脉高压(pulmonary arterial hypertension,PAH)为主要表现。PAH是MCTD严重的并发症之一,起病隐匿,易漏诊,晚期患者可出现右心功能衰竭,甚至死亡,治疗效果差[1]。目前国内外报道较少,特别是经右心导管检查确诊的病例。本文旨在分析混合性结缔组织病相关肺动脉高压(MCTD-PAH)患者的临床特点及发病危险因素,为其早期识别提供依据。
1 对象与方法
1.1 研究对象 回顾性纳入2009年9月—2022年9月在天津医科大学总医院经右心导管检查诊断的12例MCTD-PAH患者,男1例,女11例。根据性别、年龄按1∶3随机抽取同期住院的36例混合性结缔组织病无肺动脉高压(MCTD-non-PAH)患者作为对照组。所有患者均符合Alarcon-Segovia及Kahn的MCTD分类标准[2]。右心导管诊断PAH标准:静息状态下平均肺动脉压(mean pulmonary artery pressure,mPAP)≥20 mmHg(1 mmHg=0.133 kPa),肺血管阻力(pulmonary vascular resistance,PVR)≥3 wood units(WU),肺小动脉楔压(pulmonary arteriole wedge pressure,PAWP)≤15 mmHg[3]。对照组排除PAH标准:超声心动图提示的肺动脉收缩压≥ 30 mmHg,右心房(right atrium,RA)及右心室(right ventricular,RV)扩大,室间隔形状及运动异常等支持肺高压(pulmonary hypertension,PH)的表现。排除其他可能导致PH,如合并左心疾病、心脏瓣膜病、慢性阻塞性肺疾病、严重的ILD、门静脉高压、睡眠呼吸暂停综合征及肺血管栓塞等。
1.2 研究方法
1.2.1 收集指标 收集并记录2组患者年龄、性别、就诊时间、临床症状、心功能分级;相关实验室检查指标(血常规、尿常规、肝功能、肾功能、心肌酶、肌钙蛋白T、免疫球蛋白、C反应蛋白、抗核抗体谱);辅助检查结果(超声心动图、肺功能、胸部CT);MCTD-PAH组6 min步行距离(6MWD)及右心导管检查结果;治疗药物(糖皮质激素、免疫抑制剂、PAH靶向治疗药物)。
1.2.2 随访 截至2023年4月,通过住院、门诊或电话随访的方式对2组患者生存状态进行随访记录。
1.3 统计学方法 采用SPSS 22.0软件进行数据处理,正态分布的计量资料以[[x] ±s
]表示,组间比较采用独立样本t检验。非正态分布的计量资料以M(P25,P75)表示,组间比较采用秩和检验;计数资料以例(%)表示,组间比较采用卡方检验或Fisher确切概率法。危险因素筛选采用非条件Logistic回归分析。P<0.05为差异有统计学意义。
2 结果
2.1 MCTD-PAH患者临床特点 12例患者年龄25~60岁,平均(42.83±11.24)岁,7例(58.3%)以PAH症状首次就诊于心脏科,同时诊断MCTD及PAH;5例(41.7%)PAH症状晚发于MCTD表现,中位发病时间为2.0(0.5,4.0)年。主要临床症状为活动后气短10例(83.3%),乏力6例(50.0%),胸痛2例(16.7%),心悸2例(16.7%),咳嗽1例(8.3%)。诊断PAH时8例(66.7%)WHO Fc-Ⅱ级,4例(33.3%)WHO Fc-Ⅲ级,6MWD平均(359.4±79.1)m。
2.2 MCTD-PAH患者右心导管检查结果 12例患者mPAP为(38.92±7.39)mmHg,mRAP为(4.67±2.67)mmHg,PVR(8.01±3.48)WU,心指数(cardiac index,CI)为(3.05±0.87)L·min-1·m-2。
2.3 MCTD-PAH组与对照组临床特点分析 2组患者年龄、性别比例差异无统计学意义。MCTD-PAH组较对照组出现活动后气短、肌炎及心包积液比例更高,血沉、免疫球蛋白G(IgG)、RA横径、RV横径及PASP水平更高,见表1。
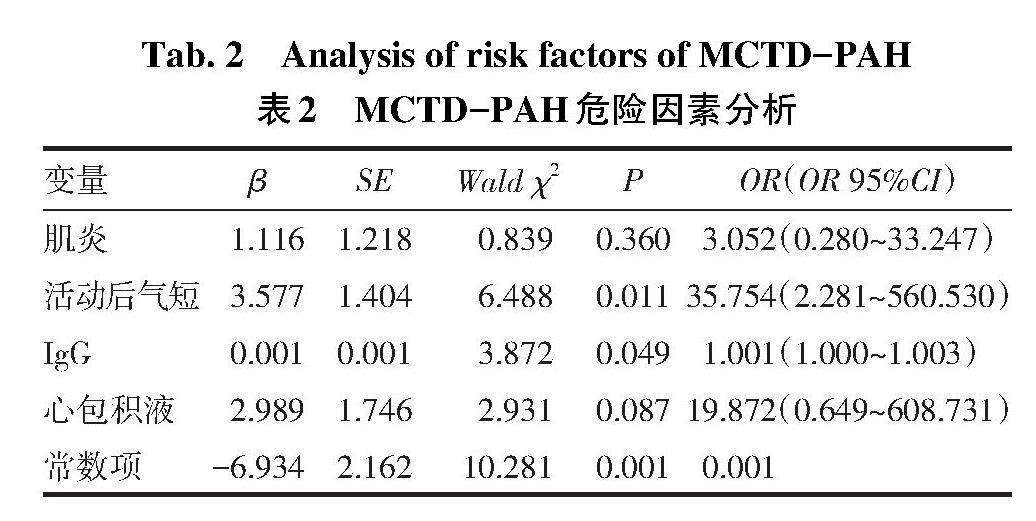
2.4 MCTD-PAH发病危险因素分析 以PAH为因变量(无PAH=0,有PAH=1),以表1中组间差异有统计学意义的指标为自变量(其中分类变量肌炎、活动后气短和心包积液均将有赋值为1,无赋值为0;因RA、RV及PASP是明确PAH相关的指标,且本文在进行PAH筛选病例时用到了这3项指标,故不再纳入多因素分析;因高IgG水平与血沉增快有关,故未纳入血沉指标),进行二元Logistic回归分析。结果显示活动后气短及较高水平的IgG是预测MCTD发生PAH的危险因素,见表2。[变量 β SE Wald χ2 P OR(OR 95%CI) 肌炎 1.116 1.218 0.839 0.360 3.052(0.280~33.247) 活动后气短 3.577 1.404 6.488 0.011 35.754(2.281~560.530) IgG 0.001 0.001 3.872 0.049 1.001(1.000~1.003) 心包积液 2.989 1.746 2.931 0.087 19.872(0.649~608.731) 常数项 -6.934 2.162 10.281 0.001 0.001 ][Tab.2 Analysis of risk factors of MCTD-PAH
表2 MCTD-PAH危险因素分析]
2.5 预后 截至2023年4月,中位随访时间56(24~84)个月。12例中9例存活,其中3例经治疗后PAH完全缓解;3例死亡,2例死因为右心功能衰竭,1例死因不详。36例MCTD-non-PAH患者均存活。
3 讨论
PAH是指由多种病因和不同的发病机制所致肺动脉结构和功能的改变,引起肺血管阻力和肺动脉压力升高的病理生理综合征,重者可发展成右心功能衰竭甚至死亡。诊断PAH时需考虑潜在系统性疾病的可能,结缔组织病(CTD)是引起PAH常见的病因之一,西方国家以系统性硬化症(SSc)最多,我国以系统性红斑狼疮(SLE)和原发性干燥综合征(pSS)多见,其次为SSc及MCTD[4]。PAH临床症状无特异性,可与MCTD的临床表现相混淆,易出现漏诊,了解MCTD-PAH的临床特点及发病危险因素对于疾病的早期识别、早期治疗及改善预后至关重要。
3.1 MCTD-PAH的临床特点 PAH患者的临床症状轻重不一,主要表现为心功能不全引起的活动后气短、周身乏力、心悸、胸痛及咳嗽,重者可出现黑曚及晕厥,少数患者可因肺动脉增粗压迫喉返神经出现声音嘶哑。上述症状无特异性,可与MCTD的临床表现相混淆。本研究中2组均可出现活动后气短、乏力、胸痛等临床表现,但MCTD-PAH组活动后气短较对照组比例更高,且是预测PAH发病的独立危险因素,提示临床医生需重视患者主诉,MCTD患者出现活动后气短时,除了需要筛查常见的ILD损害,还需警惕合并PAH。
3.2 MCTD-PAH发病危险因素 MCTD-PAH发病机制尚无明确报道,本研究发现MCTD患者诊断PAH时常合并肌炎及心包积液,血沉更快,IgG更高,多因素分析显示更高水平的IgG是MCTD-PAH发病的危险因素。在CTD-PAH中,心包积液可能由CTD引起的心包炎所致,亦可能与右心功能衰竭相关,被认为是SLE-PAH[5]及pSS-PAH[6]患者发病的危险因素。本研究显示MCTD-PAH组合并心包积液比例更高,但在多因素分析未能获得统计学意义,可能是因为本研究临床样本量较少,扩大研究样本量也许能获得阳性结果。
目前尚无临床研究报道CTD的肌炎表现与PAH发病有关,但在SSc-PAH的研究中,认为合并肌炎是患者预后差的危险因素[7],具体机制有待进一步探究。多项临床研究表明抗U1RNP抗体、抗SSA抗体、抗Ro52抗体、抗SSB抗体是SLE-PAH、pSS-PAH及SSc-PAH发病的危险因素[5-6,8]。本研究比较了2组患者的自身抗体,差异无统计学意义,CTD-PAH的发病与自身抗体的关系有待更细致深入的基础实验探究。
MCTD起病时常表现为血沉增快及高IgG血症,提示患者的炎症免疫状态活跃。有研究发现CTD-PAH患者肺血管管壁有IgG及补体的沉积,推测异常的免疫炎症反应参与了PAH患者肺血管管壁的重塑[9]。本研究分析得出更高水平的IgG血症是MCTD-PAH发病的独立危险因素,支持该假说。
3.3 MCTD-PAH预后 既往研究报道MCTD患者20年的生存率为90%[10]。MCTD-PAH患者的生存率与SSc-PAH患者相似,1年及3年的生存率为89%及63%,PAH引起的右心功能衰竭是患者死亡的重要原因[11]。本组中3例患者死亡,有2例在PAH诊断后1年内死亡,1例于PAH诊断的第4年死亡。临床研究发现MCTD-PAH患者对于激素及免疫抑制剂治疗敏感,在免疫抑制剂的选择上推荐环磷酰胺、吗替麦考酚酯、他克莫司及环孢素,少部分患者仅接受激素及免疫抑制剂治疗即可实现原发病及PAH的病情缓解[12-13]。本研究有3例患者经足量激素及环磷酰胺治疗后PAH病情缓解,停用PAH靶向药物后病情持续缓解,证实了原发病治疗在MCTD-PAH患者诊疗中的重要地位。
综上所述,免疫炎症反应可能参与了MCTD-PAH的发病。对于出现活动后气短,IgG明显升高的MCTD患者应注意排查PAH,以便早期诊断。激素及免疫抑制剂对于MCTD-PAH患者的治疗至关重要。
参考文献
[1] GUNNARSSON R,ANDREASSEN A K,MOLBERG ?,et al. Prevalence of pulmonary hypertension in an unselected,mixed connective tissue disease cohort:results of a nationwide,Norwegian cross-sectional multicentre study and review of current literature[J]. Rheumatology(Oxford),2013,52(7):1208-1213. doi:10.1093/rheumatology/kes430.
[2] 莫颖倩,严青,叶霜,等. 未分化结缔组织病和混合性结缔组织病的诊疗规范[J]. 中华内科杂志,2022,61(10):1119-1127. MO Y Q,YAN Q,YE S,et al. Standardized diagnosis and treatment of undifferentiated connective tissue disease and mixed connective tissue disease[J]. Chin J Intern Med,2022,61(10):1119-1127. doi:10.3760/cma.j.cn112138-20220104-00009.
[3] HUMBERT M,KOVACS G,HOEPER M M,et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension[J]. Eur Heart J,2022,43(38):3618-3731. doi:10.1093/eurheartj/ehac237.
[4] NIKLAS K,NIKLAS A,MULAREK-KUBZDELA T,et al. Prevalence of pulmonary hypertension in patients with systemic sclerosis and mixed connective tissue disease[J]. Medicine(Baltimore),2018,97(28):e11437. doi:10.1097/MD.0000000000011437.
[5] QU J,LI M,WANG Y,et al. Predicting the risk of pulmonary arterial hypertension in systemic lupus erythematosus:a Chinese systemic lupus erythematosus treatment and research group cohort study[J]. Arthritis Rheumatol,2021,73(10):1847-1855. doi:10.1002/art.41740.
[6] WANG J,LI M,WANG Q,et al. Pulmonary arterial hypertension associated with primary Sjogren's syndrome:a multicentre cohort study from China[J]. Eur Respir J,2020,56(5):1902157. doi:10.1183/13993003.02157-2019.
[7] FOOCHAROEN C,NANAGARA R,KIATCHOOSAKUN S,et al. Prognostic factors of mortality and 2-year survival analysis of systemic sclerosis with pulmonary arterial hypertension in Thailand[J]. Int J Rheum Dis,2011,14(3):282-289. doi:10.1111/j.1756-185X.2011.01625.x.
[8] NUNES J P L,CUNHA A C,MEIRINHOS T,et al. Prevalence of auto-antibodies associated to pulmonary arterial hypertension in scleroderma - a review[J]. Autoimmun Rev,2018,17(12):1186-1201. doi:10.1016/j.autrev.2018.06.009.
[9] FRID M G,THURMAN J M,HANSEN K C,et al. Inflammation,immunity,and vascular remodeling in pulmonary hypertension;evidence for complement involvement?[J]. Glob Cardiol Sci Pract,2020,2020(1):e202001. doi:10.21542/gcsp.2020.1.
[10] VEGH J,SZODORAY P,KAPPELMAYER J,et al. Clinical and immunoserological characteristics of mixed connective tissue disease associated with pulmonary arterial hypertension[J]. Scand J Immunol,2006,64(1):69-76. doi:10.1111/j.1365-3083.200 6.01770.x.
[11] CONDLIFFE R,KIELY D G,PEACOCK A J,et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era[J]. Am J Respir Crit Care Med,2009,179(2):151-157. doi:10.1164/rccm.200806-953OC.
[12] SANCHEZ O,SITBON O,JAIS X,et al. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension[J]. Chest,2006,130(1):182-189. doi:10.1378/chest.130.1.182.
[13] 张晓,赵久良,丁峰,等. 结缔组织病相关肺动脉高压诊疗规范[J]. 中华内科杂志,2022,61(11):1206-1216. ZHANG X,ZHAO J L,DING F,et al. Recommendations for the diagnosis and treatment of connective tissue disease associated pulmonary arterial hypertension in China[J]. Chin J Intern Med,2022,61(11):1206-1216. doi:10.3760/cma.j.cn112138-20220309-00164.
(2023-11-17收稿 2024-01-08修回)
(本文编辑 李鹏)
猜你喜欢
现代商贸工业(2016年9期)2017-01-07
中国医药导报(2016年29期)2016-12-27
中国科技纵横(2016年17期)2016-11-30
中国实用医药(2016年27期)2016-11-30
中外医学研究(2016年28期)2016-11-28
中国现代医生(2016年25期)2016-11-19
中国现代医生(2016年23期)2016-11-15
中国实用医药(2016年21期)2016-08-19
中国实用医药(2016年9期)2016-05-17
饮食与健康·下旬刊(2016年7期)2016-05-10
