
抗菌药普托马尼中3个工艺杂质的(Q)SAR遗传毒性评价与气相色谱-串联质谱法测定
2024-05-03 13:33:14钱叶飞鲁辉许奇吴杨黄逸文
中国抗生素杂志 2024年2期
钱叶飞 鲁辉 许奇 吴杨 黄逸文


摘要:目的 对新型抗结核分枝杆菌药普托马尼各合成路线涉及的3个共性工艺杂质:(S)-叔丁基二甲基硅烷缩水甘油醚(杂质I)、(S)-丁酸缩水甘油酯(杂质II)、4-三氟甲氧基溴苄(杂质III)进行遗传毒性评价并建立相应的质量控制方法。方法 分别采用基于专家规则和统计学原理的2种互补的(定量)构效关系[(Q)SAR]模型(Derek和Sarah)对普托马尼中3个工艺杂质的遗传毒性进行评价和分类;根据评价结果建立气相色谱-串联质谱(GC-MS/MS)法,采用分时段多反应监测(MRM)模式同时对这3个工艺杂质进行测定,并阐述这3个工艺杂质在EI源下的质谱裂解规律。结果 杂质I和杂质III Derek评估结果均为阳性,Sarah评估结果分别为模棱两可和阴性,依据ICH M7指南分类为3类致突变杂质;杂质II Derek评估结果为阳性,Sarah结果显示有明确的Ames阳性实验结果,为2类已知致突变杂质。3个工艺杂质均需按照毒理学关注阈值(TTC)进行控制,建立的GC-MS/MS法经验证3个杂质可有效分离,线性关系良好,方法定量限均低于拟定限度的15%,平均回收率(n=9)分别为105.5%、104.4%和108.5%,重复性RSD(n=6)分别为2.2%、5.8%和2.2%。3批样品均检出杂质III。结论 建立的GC-MS/MS法操作简单,专属性强,灵敏度高,可用于普托马尼中3个潜在致突变杂质的测定。由于杂质II也是恶唑烷类抗菌药如利奈唑胺、咔哒唑胺、泰地唑胺等的共性工艺杂质,因此本研究也为其他恶唑烷類抗菌药中杂质II的质量控制提供了参考。
关键词:普托马尼;遗传毒性杂质;(定量)构效关系;气相色谱-串联质谱法
中图分类号:R978文献标志码:A
(Q)SAR genotoxicity evaluation and GC-MS/MS determination of three process impurities in pretomanid, an antibacterial agent
Abstract Objective To evaluate the genotoxicity of three process impurities involved in various synthetic routes of pretomanid, a new antimycobacterial drug, and to establish the quantitative method of these three impurities, namely (S)-tert-butyldimethyl(oxiran-2-ylmethoxy) silane (impurity I), (S)-oxiran-2-ylmethyl butyrate (impurity II), and 1-(bromomethyl)-4-(trifluoro methoxy)benzene (impurity III). Methods Two complementary (quantitative) structure-activity relationship [(Q)SAR] evaluation models (Derek and Sarah) based on expert rules and statistics, respectively, were employed to assess and classify the genotoxicity of the three process impurities in pretomanid, and a GC-MS/MS method with the time-segmented multiple reaction monitoring (MRM) mode was subsequently developed for the simultaneous determination of the three impurities. The fragmentation patterns of the three impurities in EI source were also discussed in this study. Results The Derek evaluation results were all positive for impurities I and III, while the Sarah results were equivocal and negative, respectively, indicating impurities I and III were categorized as class 3 as per ICH M7 guideline. Impurity II was regarded as a confirmed mutagenic impurity of class 2 since Sarah showed specific positive results in Ames test. The threshold of toxicological concern (TTC) was applied to control these three impurities. The developed GC-MS/MS method was validated and showed effective resolutions between the impurities with good linearity. The LOQ values of the three impurities were all as low as 15% of the acceptable limit. The average recoveries (n=9) were 105.5%, 104.4%, and 108.5%, while the repeatabilities RSD (n=6) were 2.2%, 5.8%, and 2.2%, respectively. Impurity III was detected in all batches. Conclusion The established method is easy to operate and proved selective and sensitive, which is applicable for quality control of the three potential genotoxic impurities in pretomanid. This study can also be referenced for the quality control of impurity II in other oxazolidinone antibacterial drugs like linezolid, cadazolid, and tedizolid, as impurity II was the common impurity.
Key words Pretomanid; Genotoxic impurity; (Q) SAR; GC-MS/MS
普托馬尼(pretomanid, PA-824),化学名为(S)-2-硝基-6-[4-(三氟甲氧基)苄氧基]-6,7-二氢-5H-咪唑并[2,1-b] [1,3]恶嗪,是一种硝基咪唑并恶嗪类的新型抗结核分枝杆菌药物,由全球结核病药物开发联盟开发,于2019年8月获FDA批准上市,与贝达喹啉(bedaquiline)和利奈唑胺(linezolid)联用,用于治疗特定类型的高度耐药肺结核患者[1-2]。普托马尼是近40年来FDA批准的第3个抗肺结核新药,也是第1个由非营利组织开发上市的抗肺结核新药[3-4]。
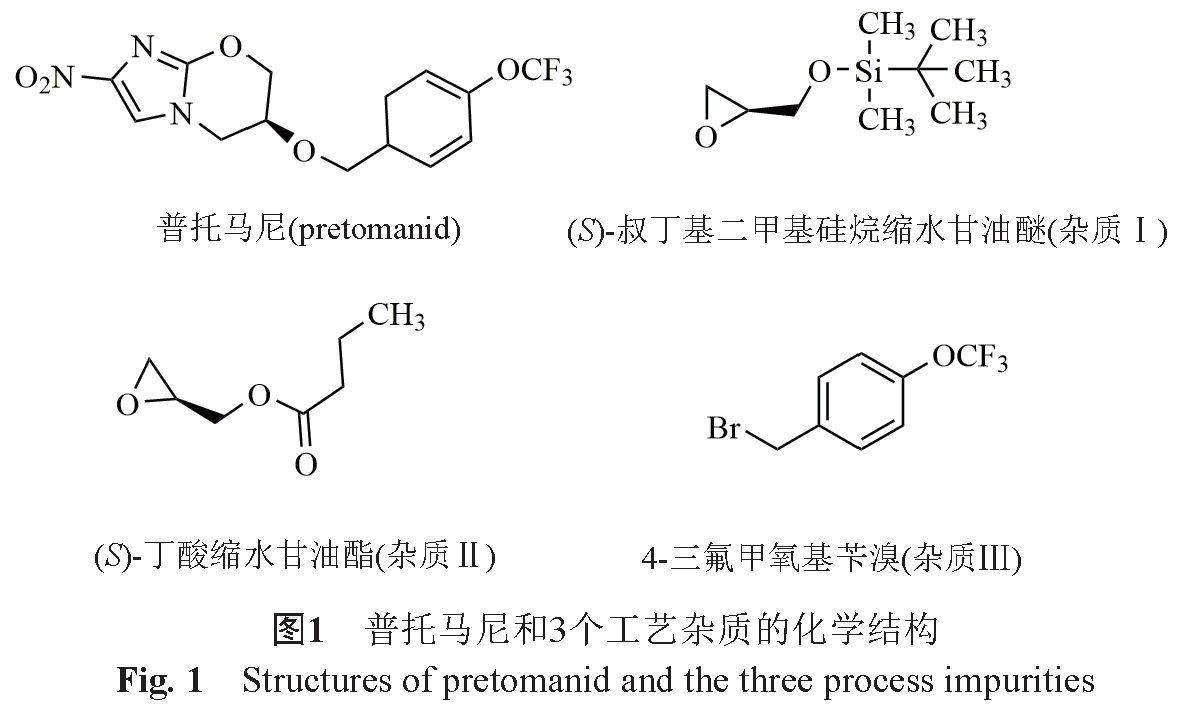
普托马尼的生产工艺涉及多种合成路线[5-7],但其核心均为构建硝基咪唑并恶嗪环的手性中心以及引入4-三氟甲氧基苄基侧链。其中手性中心的构建多是通过带有保护基的(S)-缩水甘油醇作为手性源直接引入,主要为(S)-叔丁基二甲基硅烷缩水甘油醚和(S)-丁酸缩水甘油酯;而侧链均通过4-三氟甲氧基苄溴引入。因此,(S)-叔丁基二甲基硅烷缩水甘油醚、(S)-丁酸缩水甘油酯和4-三氟甲氧基苄溴可作为普托马尼各合成路线的共性工艺杂质残留在终产品中,且分别含有典型的遗传毒性警示结构—环氧结构和卤代烷烃结构[8-9]。普托马尼和3个工艺杂质的化学结构见图1。
作为已获批上市的药物,普托马尼本身的毒性包括遗传毒性相关信息是已知和公开的,其已通过体外细菌回复突变试验(Ames)证明无致突变性[10]。然而,药物杂质的毒性相关信息通常是不公开或未知的。此外,人用药品技术要求国际协调理事会(ICH) M7(R1)指南(《评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险》)提出,对于药物杂质应关注其遗传毒性,尤其是致突变性,对于非致突变性杂质可按普通杂质进行控制,并明确提出不应仅依据目视法有无警示结构而判定是否为遗传毒性(致突变性)杂质[11-12]。
由于对所有药物杂质进行体内外相关毒理实验是不现实的,(定量)构效关系[(Quantitative) Structure-Activity Relationships, (Q)SAR]技术作为一种通过化合物现有资料、化学结构和对Ames试验结果的预测而评估其致突变性的计算机毒理学模型[13-15],被各监管机构和指南广泛推荐用于药物杂质的遗传毒性(致突变性)的评估和分类[11-12]。ICH M7指南要求采用2种互补的(Q)SAR评估模型,一种基于专家规则,一种基于统计学原理,且对于分类为2类或3类的致突变杂质,应采用毒理学关注阈值(TTC,1.5 μg/d)作为每日可接受摄入量并严格控制[11]。普托马尼每日最高服用剂量为0.2 g,因此其中的致突变杂质限度为7.5 μg/g,对分析方法的灵敏度提出了较高的要求。
目前尚无普托马尼相关杂质毒性评价及这3个工艺杂质测定的研究报道[16-17],本研究分别采用基于专家规则的Derek模型和统计学原理的Sarah模型这2种(Q)SAR工具对普托马尼中的这3个共性工艺杂质进行遗传毒性评估与分类,并根据评估结果建立了专属灵敏的气相色谱-串联质谱(GC-MS/MS)法,采用分时段多反应监测(MRM)模式同时对这3个工艺杂质进行测定。此外,本研究对这3个工艺杂质在EI源下的质谱裂解规律也进行了阐述。
1 仪器与试药
1.1 (Q)SAR模型
Derek版本:Derek Nexus 6.1.1;数据库:Derek KB 2020 1.0;数据库版本:1.0;数据库日期:2020年3月26日。Sarah版本:Sarah Nexus 3.1.1;模型:Sarah Model 2020.1;模型版本:1.8。Nexus版本(Derek和Sarah的整合平台):Nexus 2.4.0。以上系统均由英国Lhasa公司(http://www.lhasalimited.org/)开发。
1.2 仪器
8890-7010B型气相色谱质谱联用仪(美国Agilent公司);XSE205DU型十万分之一电子分析天平(瑞士Mettler公司)。
1.3 试剂与试药
(S)-叔丁基二甲基硅烷缩水甘油醚对照品(批号:7M4BH,纯度:99.7%)、(S)-丁酸缩水甘油酯对照品(批号:UXB5A,纯度:98.2%)和4-三氟甲氧基苄溴对照品(批号:S7JUE,纯度:99.1%)均购自东京化成工业株式会社(TCI);3批普托马尼样品(批号:PA20201001、PA20210321、PA20210506)由烟台药物研究所提供;色谱级四氢呋喃购自美国Sigma公司。
2 方法
2.1 (Q)SAR遗传毒性评价
采用Chemdraw绘制普托马尼和3个杂质的化学结构后导入Nexus 2.4.0系统,物种设为细菌(bacterium),预测终点设置为遗传毒性(genotoxicity)项下的致突变性(mutagenicity)。采用ICH M7分类模式(ICH M7 classification)对遗传毒性进行评价和自动分类。
2.2 色谱条件
采用Agilent HP-5MS色谱柱(30 m×0.25 mm, 0.25 μm);载气为氦气,流速1.0 mL/min;进样口温度200 ℃;分流进样,分流比5∶1;进样体积1 μL;柱温为程序升温(起始温度60 ℃,保持1 min,以8 ℃/min升温至130 ℃,保持1 min,再以60 ℃/min升温至250 ℃,保持5 min)。
2.3 质谱条件
采用EI源,电子能量70 eV;离子源温度230 ℃;质谱传输线温度250 ℃;扫描模式为分时段MRM;各化合物监测时间段、定性与定量离子对及对应的碰撞能详见表1;驻留时间(dwell time)均为80 ms;溶剂延迟时间5 min;运行时间12 min;增益因子为10。
2.4 溶液制备
空白溶剂:四氢呋喃
对照品贮备液:分别精密称取杂质I、杂质II、杂质III对照品适量,置同一量瓶中,加溶剂溶解并稀释制成浓度均为100 ng/mL的混合溶液。
线性考察溶液:精密量取对照品贮备液适量,加溶剂逐级稀释制成浓度分别为5、10、20、40、80和100 ng/mL的系列溶液。
对照品溶液:精密量取对照品贮备液适量,加溶剂稀释制成浓度为40 ng/mL的溶液。
供试品溶液:精密称取普托马尼样品适量,加溶剂溶解制成5 mg/mL的溶液。
加样回收率溶液:分别精密称取普托马尼样品(批号:PA20210321)适量,置9个量瓶中,分别加入5、40和80 ng/mL的系列对照品溶液,溶解制成低、中、高3个浓度水平的加样回收率溶液,每个浓度水平平行制备3份。
重复性考察溶液:精密称取普托马尼样品(批号:PA20210321)适量,加对照品溶液溶解制成100%浓度水平的加标供试品溶液,平行制备6份。
3 结果
3.1 (Q)SAR遗传毒性评价
Derek是通过将用户输入结构与专家知识库中的警示结构规则比对,然后突出显示所有和毒性相关的警示结构,给出毒性评估结果;而Sarah是基于机器学习算法,从Ames实验数据出发,构建统计模型。依据ICH M7指南,如果这2种互补的计算机评价模型均给出致突变性为阴性的预测结果,可以认为该杂质无致突变性,归类为5类普通杂质;相反,若2种软件中至少有1种给出阳性预测结果,则认为该杂质致突变性为阳性,归类为3类;若有明确的致癌性或致突变性数据(非预测),则进一步归类为1类或2类。此外,若药物本身即存在相同的警示结构,则杂质分类为4类,也按照非致突变杂质即普通杂质控制。
结果如表2所示,杂质I和杂质III Derek评估结果均为阳性,显示的警示结构分别为环氧化物和烷化剂,且这2类警示结构未存在于普托马尼结构中;Sarah评估结果分别为模棱两可和阴性(置信度47%),杂质I和杂质III最终分类为3类致突变杂质。杂质II Derek评估结果为阳性,显示的警示结构为环氧丙基酯,且该警示结构未存在于普托马尼结构中;Sarah结果显示为100%阳性,即杂质II有明确的体外Ames阳性实验结果,为2类已知致突变杂质。3个工艺杂质均需按照TTC进行控制。
3.2 质谱裂解规律与离子对的选择
取混合对照品贮备液在m/z 40~400范围内进行全扫描(full scan),由图2各杂质一级质谱数据可知,在EI源标准70 eV电离能下,除杂质III可见很弱的分子离子峰m/z 254,杂质I和杂质II均未见分子离子峰。其中杂质I的一级质谱图中最高质量数为m/z 131,为基于醚类化合物α断裂的产物;杂质II一级质谱图中最高质量数为m/z 116,为γ-H发生McLafferty重排(麦氏重排)后脱去一分子乙烯的产物;杂质III虽可见分子离子峰m/z 254,但丰度太低,影响定量灵敏度,第二大质量数为m/z 175,为i断裂脱溴自由基的产物,且丰度最高。由于较高的质量数结构特异性和抗干扰能力强,同时兼顾丰度,最终选择m/z 131、116、175作为3个杂质的定量母离子。
进一步对各化合物母离子设置不同碰撞能(CE)进行子离子扫描(图3)。结果显示,作为有机硅化物的特有裂解规律[18-19],杂质I的母离子m/z 131可分别经烷基重排脱甲醛裂解以及经H重排脱醛裂解生成碎片离子m/z 101和59;同时作为醚类化合物母离子m/z 131亦可发生四元环过渡态重排生成碎片离子m/z 75(图4A)。其中m/z 101丰度最高且在10 eV碰撞能下达到峰值,因此最终以m/z 131→m/z 101作为定量离子对,CE设为10 eV。将丰度第二高的m/z 59作为定性子离子,CE经筛选为25 eV。
雜质II的母离子m/z 116通过α断裂生成碎片离子m/z 43;或通过i断裂生成碎片离子m/z 42和57(图4B)。其中m/z 57丰度最高且在10 eV碰撞能下达到峰值,因此以m/z 57作为定量离子对,CE设为10 eV。定性子离子设为m/z 42,CE为18 eV。
杂质III的母离子m/z 175经中性丢失生成三氟碳正离子m/z 69;或经苯环扩环形成稳定的?鎓离子,失去一分子环戊二烯后生成碎片离子m/z 109(图4C)。其中m/z 109丰度最高,因此最终以m/z 175→m/z 109作为定量离子对,以m/z 175→m/z 69为定性离子对。
3.3 方法学验证
3.3.1 专属性
分别取“2.4”项下空白溶液、对照品溶液、供试品溶液(批号:PA20210321),按“2.2”和“2.3”项下色谱与质谱条件进样分析,记录色谱图(图5),结果表明空白溶液和样品基质不干扰3个目标化合物的测定。
3.3.2 线性与范围
取“2.4”项下系列线性考察溶液分别进样,记录色谱图,以浓度(x)为横坐标,峰面积(y)为纵坐标按最小二乘法进行回归分析(n=6),结果见表3。3个目标化合物在5~100 ng/mL范围内质量浓度与响应值线性关系显著(P<0.001)。且截距95%置信区间均包含0,说明截距与0无显著性差异(P>0.05),可采用外标一点法计算各目标化合物含量。
3.3.3 检测限与定量限
用溶剂将对照品溶液逐级稀释,按“2.2”和“2.3”项下色谱与质谱条件进样分析,记录色谱图。以信噪比>10时相应的浓度作为定量限(LOQ)浓度,以信噪比>3时相应的浓度作为检测限(LOD)浓度。结果详见表3,3个杂质LOQ浓度低于限度浓度的15%,表明方法灵敏度满足限度要求。LOQ连续进样6次,RSD(n=6)分别为2.7%、8.3%和1.7%,表明LOQ精密度良好。
3.3.4 回收率
取“2.4”项下加样回收率溶液,按“2.2”和“2.3”项下色谱与质谱条件进样测定,计算各目标化合物在LOQ、100%、200% 3个浓度水平的平均回收率,结果见表4。
3.3.5 重复性
取“2.4”项下6份重复性考察溶液,按“2.2”和“2.3”项下色谱与质谱条件进样测定,结果各目标化合物峰面积RSD(n=6)分别为2.2%、5.8%和2.2%,表明方法重复性良好。
3.3.6 溶液稳定性
取“2.4”项下对照品溶液在室温下分别于0、4、8、12和16 h进样测定,结果各目标化合物峰面积RSD(n=5)分别为2.5%、7.7%和1.7%,表明溶液在室温下16 h内稳定性良好。
3.4 样品测定
取3批普托马尼样品制备供试品溶液后进样测定,按外标法计算3个遗传毒性杂质的含量,结果显示3批样品均检出杂质III,含量分别为1.3 μg/g、 4 讨论 经溶剂筛选发现普托马尼在甲醇和四氢呋喃中溶解度较好,适合制备较高浓度的供试品溶液来提高方法灵敏度,其中以四氢呋喃为溶剂时目标峰信号响应更强,因此作为样品溶剂。由于3个杂质极性较小,因此选择了弱极性柱HP-5MS作为色谱柱,3个化合物均有较好的保留,在full scan模式下经程序升温的优化,各化合物之间以及与空白峰互不干扰。杂质I和杂质II均为环氧化物,在高温下稳定性较差,易开环降解,但较低的进样口温度不利于目标化合物的挥发和检测,筛选了180、200、220和250 ℃等不同进样口温度,通过在full scan模式下观察目标峰响应和其他干扰峰的大小,最终选择进样口温度为200 ℃。根据3个目标化合物出峰时间,选择了分时间段的MRM监测模式,使得在一定的总扫描时间内,各化合物的离子对通道被分配更长的驻留时间,从而提高方法灵敏度。各化合物母离子、子离子以及碰撞能等质谱参数的选择详见“3.2”项下。 作为新型抗结核分枝杆菌药,普托马尼的合成工艺有较多文献报道,本研究基于不同合成策略涉及到的3个共性工艺杂质,采用Derek和Sarah 2个原理互补的商业化(Q)SAR模型对其进行了遗传毒性(致突变性)评价;并根据阳性评估结果建立了专属、灵敏的GC-MS/MS测定法,依据3个杂质的裂解规律选择合适的定量离子对和CE提高方法选择性和灵敏度,并采用分时段的MRM监测模式进一步提高检测灵敏度。结果3批样品均检出杂质III。此外,由于杂质II也是利奈唑胺、咔哒唑胺、泰地唑胺等恶唑烷类抗菌药的常用手性合成砌块,因此本研究也为其他恶唑烷类抗菌药中杂质II的质量控制提供了参考。 參 考 文 献 Gils T, Lynen L, de Jong B C, et al. Pretomanid for tuberculosis: A systematic review[J]. Clin Microbiol Infect, 2022, 28(1): 31-42. Fekadu G, Tolossa T, Turi E, et al. Pretomanid development and its clinical roles in treating tuberculosis[J]. J Glob Antimicrob Resist, 2022, 31: 175-184. 王玉丽, 张洪兵, 刘昌孝, 等. 回顾分析:2011—2020年美国批准上市的抗感染药物[J]. 中国抗生素杂志, 2022, 47(1): 1-14. Keam S J. Pretomanid: First approval[J]. Drugs, 2019, 79(16): 1797-1803. 高磊, 杨德志. 普托马尼的合成研究进展[J]. 中国医药工业杂志, 2021, 52(4): 463-470. 黄娟, 龙超久. 普瑞玛尼合成路线图解[J]. 国外医药(抗生素分册), 2020, 41(6): 482-485. Flick A C, Leverett C A, Ding H X, et al. Synthetic approaches to the new drugs approved during 2019[J]. J Med Chem, 2021, 64(7): 3604-3657. Snodin D J. A primer for pharmaceutical process development chemists and analysts in relation to impurities perceived to be mutagenic or "Genotoxic"[J]. Org Process Res Dev, 2020, 24(11): 2407-2427. 吴杨, 张强, 许奇, 等. 甲磺酸伏美替尼中间体AST 2815中4个基因毒性杂质检测[J]. 药物分析杂志, 2022, 42(7): 1170-1177. FDA. Drugs@FDA: FDA-approved drugs[EB/OL]. [2019-08-14]. https://www.accessdata.fda.gov/drug satfda_docs/label/2019/212862s000lbl.pdf. ICH. ICH Guideline-Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk M7(R1)[S]. 2017. ICH. ICH M7 Q&As, Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potentialcarcinogenic risk questions & answers[S]. 2020. 陈莉, 祝清芬, 季文君, 等. 色瑞替尼有关物质遗传毒性(Q)SAR评价及Ames试验研究[J]. 中国现代应用药学, 2021, 38(24): 3082-3086. 祝清芬, 王维剑. (Q)SAR技术在药物杂质遗传毒性评价中的应用[J]. 中国药物评价, 2021, 38(5): 371-374. Hasselgren C, Bercu J, Cayley A, et al. Management of pharmaceutical ICH M7 (Q)SAR predictions-the impact of model updates[J]. Regul Toxicol Pharmacol, 2020, 118: 104807. Surapuraju P K R, Juturu R R. Development and validation of stability-indicating-HPLC method for the determination of related substances in novel nitroimidazole antituberculosis drug pretomanid: Robustness study by Design-Expert and application to stability studies[J]. Biomed Chromatogr, 2022, 36 (12): e5498. Momin M A M, Thien S J, Krittaphol W, et al. Simultaneous HPLC assay for pretomanid (PA-824), moxifloxacin and pyrazinamide in an inhaler formulation for drug-resistant tuberculosis[J]. J Pharm Biomed Anal, 2017, 135: 133-139. 林吉茂, 周愛民, 李慧惠. 二甲基二烷氧基硅烷质谱研究[J]. 结构化学, 1995, 14(Z1): 412-416. 王姗姗, 蒋可志, 伍川. 气质联用技术在有机硅化合物分析中的应用[J]. 杭州师范大学学报(自然科学版), 2014, 13(6): 579-585.
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中成药(2018年12期)2018-12-29 12:25:44
中成药(2017年6期)2017-06-13 07:30:35
分析化学(2017年1期)2017-02-06 21:34:35
中国纤检(2016年12期)2017-01-20 09:28:19
现代农业科技(2016年20期)2016-12-20 09:05:36
分析化学(2016年7期)2016-12-08 00:09:44
分析化学(2016年7期)2016-12-08 00:07:08
价值工程(2016年29期)2016-11-14 01:34:54
中国当代医药(2016年2期)2016-03-03 20:29:24
