
具有聚集诱导发光性质的铂(II)金属大环的合成
2024-04-27 09:40:06陈茂文刘森坤陈前进张灯青
合成化学 2024年4期
彭 宇, 陈茂文, 刘森坤, 陈前进, 张灯青
(东华大学 化学与化工学院,上海 201620)
铂(II)金属大环是由金属铂配体和有机配体通过配位驱动自组装[1](CDSA)所形成的离散型分子实体。通过合适的分子设计,借助金属铂原子和有机配体之间的配位作用来构筑具有特定形状和尺寸的离散型金属环。在各种自组装策略中,配位驱动自组装被认为是最有影响力的策略之一。与耗时长且产率低的分步共价合成法相比,其产率几乎为定量合成,同时不需要纯化就可以得到纯净的超分子金属环。迄今为止,科研工作者已经制备了大量二维三角形[5]、正方形[6]、矩形[7]、菱形[8]、六边形[9]和双菱形[10]等不同结构的铂(II)金属环,并且铂(II)金属环家族还在不断完善中。铂(II)金属环家族不仅在形状和尺寸上有着千差万别,而且还具有各种各样的潜在应用,比如在生物成像与癌症治疗[11]、生物传感[12]和光化学催化[13]等诸多方面有着巨大的应用前景。STANG[2]等合成了一种四苯乙烯(TPE)的铂(II)金属环,具有良好的AIE特性和与缺电子底物相互作用的能力,已成功用于硝基芳香分子的传感研究;ZHANG[3]等合成了四边形铂(II)金属环,在水溶液的C—H键烷基化光化学催化过程中表现出显著的催化活性,可应用于水溶液中光捕获体系的构建。CHEN[4]等合成了两亲性铂(II)金属环,对金黄色葡萄球菌具有显著的生物膜抑制作用,可有效缓解金黄色葡萄球菌引起的肺炎。
由于TPE单元良好的聚集诱导发光(AIE)性质,因而构建了铂(II)金属大环化合物。在稀溶液中,TPE分子产生活跃的分子内旋转,分子激发态能量以非辐射弛豫的形式耗散,导致发射被淬灭。而在聚合状态下,由于物理约束,分子内的旋转受到限制,使非辐射弛豫路径被阻断,从而激活了辐射弛豫,并产生荧光发射。因此选择TPE作为构建大环化合物的发光单元可使铂(II)金属大环具有良好的AIE性质,在荧光成像[14],固态照明[15]方面表现出良好的应用前景。
本文以4,4-二溴四苯乙烯为原料,经硝化反应得到化合物2,再将化合物2中硝基还原得到化合物3,化合物3与十二烷基酰氯发生缩合反应得到化合物4,化合物4与4-乙炔基吡啶盐酸盐发生Sonogashira偶联反应得到四苯乙烯衍生物5。5与铂(II)金属配体6通过[3+3]配位自组装合成六边形铂(II)金属大环化合物7(图1),其结构经过核磁、质谱和紫外可见吸收光谱表征。
1 实验部分
1.1 仪器与试剂
Persee Model TU-1901型紫外可见分光光度计; Brucker Model AV-400 MHz型核磁共振仪(CDCl3、 DMSO-d6或Acetone-d6为溶剂,TMS为内标); Micromass Quattro II Triple-Quadrupole型电喷雾飞行时间质谱仪。
化合物2[16]、化合物3[17]和化合物4[18]分别按参考文献方法合成。化合物1、化合物6、水合肼、钯碳加氢催化剂、十二烷基酰氯、四三苯基磷钯和三氟甲磺酸银(AgOTf)均购自上海毕得医药科技股份有限公司;4-乙炔基吡啶盐酸盐、碘化亚铜、甲苯、三乙胺、二氯甲烷、石油醚、乙酸乙酯、丙酮和四氢呋喃均购自北京百灵威科技有限公司;紫外-可见光谱测试溶剂为HPLC级,其余所用试剂均为分析纯且无需特殊处理。
1.2 合成
(1) 化合物2的合成
在冰水浴条件下,首先在250.0 mL反应瓶中加入4,4-二溴四苯乙烯(1.99 g, 4.06 mmol),乙酸(7.0 mL, 122.40 mmol),然后加入二氯甲烷(20.0 mL)作为反应溶剂。待反应体系在磁力搅拌下混合均匀,将浓硝酸(7.2 mL, 160.00 mmol)加入反应体系中。接着在25 ℃的反应温度下继续反应4 h。用水淬灭反应,再用CH2Cl2萃取混合溶液,重复3次,收集有机相,将3次收集的有机相混合后用饱和盐水溶液洗涤,随后用无水MgSO4进行干燥。使用旋转蒸发仪旋干滤液收集固体,选择硅胶柱进行纯化(洗脱剂:二氯甲烷 ∶石油醚=1∶5,V∶V)得黄色固体粉末0.94 g,产率40.0%;1H NMR(600 MHz, CDCl3)δ:8.06(d,J=8.9 Hz, 4H), 7.33(d,J=8.5 Hz, 4H), 7.17(d,J=8.7 Hz, 4H), 6.88(d,J=8.5 Hz, 4H)。
(2) 化合物3的合成
首先在100.0 mL圆底反应瓶中添加化合物2(464.00 mg, 0.80 mmol),接着加入钯碳加氢催化剂(80.00 mg, 0.08 mmol),然后进行氩气保护,再加入20.0 mL四氢呋喃(THF)溶解固体化合物,最后向反应体系中加入水合肼试剂(4.8 mL, 100.00 mmol)。随后将整个反应体系加热回流48 h。反应结束后,待反应体系自然恢复至常温,滤掉反应体系中的不溶性残留物,然后减压除去溶剂,得到棕色固体。将粗产物经硅胶柱层析纯化(洗脱剂:石油醚 ∶乙酸乙酯=4 ∶1,V∶V)得到黄色固体粉末333.00 mg,产率80%;1H NMR(600 MHz, CDCl3)δ:7.23(d,J=8.4 Hz, 4H), 6.89(d,J=8.5 Hz, 4H), 6.80(d,J=8.5 Hz, 4H), 6.45(d,J=8.5 Hz, 4H), 3.64(s, 4H);13C NMR(151 MHz, CDCl3)δ:145.13, 143.37, 142.40, 135.02, 133.64, 133.10, 132.59, 130.91, 119.88, 114.38。
(3) 化合物4的合成
首先将化合物3(500.00 mg, 0.96 mmol)加入到100.0 mL圆底烧瓶中,氩气保护,再将十二烷基酰氯(2.5 mL, 11.00 mmol)和三乙胺(0.3 mL, 2.24 mmol)加入到反应体系中,最后加入10.0 mL二氯甲烷作为反应溶剂,常温反应4 h。反应结束后,使用体积分数为10%的盐酸水溶液洗涤反应混合物,随后用饱和碳酸氢钠溶液中和,再用水(3×30 mL)洗涤混合溶液,将所得溶液使用干燥剂干燥后进行过滤。随后将滤液旋干,采用柱层析法进行纯化(洗脱剂:乙酸乙酯 ∶石油醚=1 ∶10,V∶V)得白色固体粉末603.00 mg,产率71%;1H NMR(400 MHz, CDCl3)δ:7.28(d,J=8.1 Hz, 4H), 7.22(d,J=8.2 Hz, 4H), 7.04(s, 2H), 6.9(d,J=8.3 Hz, 4H), 6.86(d,J=8.2 Hz, 4H), 2.32(t,J=7.6 Hz, 4H), 1.75~1.65(m, 4H), 1.30~1.24(m, 32H), 0.89(t,J=6.2 Hz, 6H);13C NMR(101 MHz, CDCl3)δ:171.48, 142.31, 141.02, 138.71, 137.93, 136.80, 132.96, 131.96, 131.11, 120.70, 118.99, 37.88, 31.92, 29.64, 29.62, 29.49, 29.39, 29.34, 29.31, 25.61, 22.70, 14.14; ESI-MSm/z:calcd for C50H64Br2N2O2{[M+H+]}885.3, found 885.3, {[M+Na+]}907.3, found 907.3, {[M+K+]}923.3, found 923.3。
(4) 化合物5的合成
首先将化合物4(1.77 g, 2.00 mmol), 4-乙炔基吡啶盐酸盐(1.12 g, 8.00 mmol), Pd(PPh3)4(0.23 g, 0.20 mmol),碘化亚铜(0.19 g, 1.00 mmol)和三乙胺(20.0 mL)加入到100.0 mL Schlenk瓶中,再将整个反应体系置于氩气环境中。将反应混合物在70 ℃下回流搅拌72 h。待反应结束后,将反应体系冷却至室温,然后过滤去除催化剂,用CH2Cl2(3×10 mL)反复萃取。收集有机相,使用干燥剂除去水分,减压旋干溶剂得粗产品,随后使用柱层析法(洗脱剂:二氯甲烷 ∶乙酸乙酯=10 ∶1,V∶V)进一步纯化得淡黄色固体0.93 g,产率50%;1H NMR(600 MHz, CDCl3)δ:8.58(d,J=8.4 Hz, 4H), 7.34(d,J=8.4 Hz, 4H), 7.31(d,J=8.4, 8H), 7.16(s, 2H), 7.03(d,J=8.4 Hz, 4H), 6.96(d,J=8.4 Hz, 4H), 2.32(t,J=7.6 Hz, 4H), 1.73~1.66(m, 4H), 1.37~1.23(m, 32H), 0.87(t,J=7.0 Hz, 6H);13C NMR(101 MHz, CDCl3)δ:171.39, 149.42, 144.68, 141.82, 138.58, 137.05, 132.12, 131.84, 131.58, 131.53, 125.60, 120.07, 118.92, 94.51, 87.07, 37.88, 31.91, 29.62, 29.61, 29.48, 29.39, 29.33, 29.30, 25.59, 22.69, 14.13; ESI-MSm/z:calcd for C64H72N4O2{[M+H]+}929.6, found 929.6。
(5) 六边形铂(II)金属大环化合物7的合成
将化合物5(6.27 mg, 5.0μmol), AgOTf(3.85 mg, 21.0μmol)和金属配体6(4.64 mg, 7.0μmol)溶解在丙酮和二氯甲烷混合液(3.0 mL, 1 ∶2,V∶V)中,整个反应体系置于10.0 mL棕色瓶中,并于黑暗环境中搅拌反应16 h。待反应结束后,混合液以6000 r/min离心0.5 h,收集上层清液。在上层清液中加入乙醚,出现浑浊絮状物,静置4 h后,将浑浊溶液以4000 r/min离心5 min,收集离心管中的固体,使用真空泵减压除去固体中的溶剂得到黄色固体粉末10.70 mg,产率97%;1H NMR(400 MHz, Acetone-d6)δ:9.17(s, 2H), 9.00(d,J=6.1 Hz, 4H), 7.89~7.81(m, 4H), 7.51(d,J=8.4 Hz, 4H), 7.46(d,J=8.1 Hz, 4H), 7.27(s, 1H), 7.25(d,J=5.4 Hz, 3H), 7.18(dd,J=8.3 Hz, 2.2 Hz, 4H), 6.97(d,J=8.3 Hz, 4H), 2.35(t,J=7.4 Hz, 4H), 1.98(td,J=6.6 Hz, 5.7 Hz, 2.8 Hz, 24H), 1.69~1.62(m, 4H), 1.25(dd,J=16.9 Hz, 8.3 Hz, 68H), 0.90~0.85(m, 6H);31P NMR(162 MHz, Acetone-d6)δ:15.35(s,1JPt-P=1163.12 Hz); ESI-TOF-MSm/z:calcd for C246H300F18N12O24P12Pt6S6{[M-4OTf-]4+}1551.36, found 1551.36, {[M-5OTf-]5+}1179.32, found 1179.32。
2 结果与讨论
2.1 表征
铂(II)金属大环化合物7相较于双吡啶配体化合物5,吡啶质子氢均向低场位移,图1中Ha从δ8.52移动至δ9.00, Hb从δ7.24移动至δ7.51,这归因于吡啶氮与铂配位后,其电子云密度和屏蔽效应均降低,化学位移值增加并向低场移动。31P{1NMR}在δ15.35处有一单峰,表明存在1个单一的磷环境,且相较于化合物6,化合物7的特征峰向低场移动。此外,通过ESI-TOF Mass(图2)进一步验证了大环的结构。大环化合物7的m/z{[M-4OTf-]4+}理论值为1511.38,实验值为1511.36;m/z{[M-5OTf-]5+}理论值为1179.32,实验值为1179.32。核磁和质谱数据共同证明化合物5和化合物6完成了配位自组装,成功合成了金属大环化合物7。
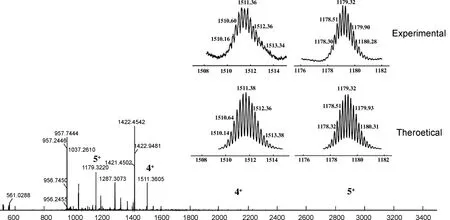
m/z
2.2 聚集诱导发光性质
图3为化合物5、化合物6和化合物7在浓度为1×10-5mol·L-1的CH2Cl2溶液中的UV-Vis谱图。化合物5的最大吸收峰出现在279 nm,这归因于四苯乙烯衍生物的吸收;化合物6最大吸收峰在302 nm;化合物7在波长302 nm处有最大吸收峰,这归因于金属到配体的电荷跃迁。化合物7在274 nm处出现肩峰,与化合物5的吸收峰接近,化合物7的吸收光谱基本上是化合物5和化合物6的叠加,说明在大环化合物7中没有形成新的共轭。

λ/nm
如图4(a)所示,铂(II)金属大环化合物7在纯DMSO溶液或在水含量较低的混合溶液中显示出较弱的荧光强度。当含水量达到30%时,荧光强度较之前有大幅度升高;含水量达到40%时,荧光强度达到最大,此时的荧光强度是化合物7在纯DMSO中荧光强度的40倍,这主要是由于提供非辐射弛豫途径的侧面苯环在聚集时运动受到限制,导致观察到显著的发射增强。当含水量超过40%时,随着水的比例不断增加,溶液的荧光强度不断下降,这可能是随着混合溶液中不良溶剂的增加,化合物7在混合溶液中溶解度下降,使快速聚集的溶质分子存在荧光屏蔽效应或者随机分子堆积形成发射较少的非晶纳米聚集体都有可能是降低荧光强度的原因[19]。从图4(b)中可以看出,随着混合溶液中含水量的不断增加,化合物7的最大发射波长出现了较为明显的蓝移现象。基于TPE的AIE机制,给定的发射波长偏移通常与外围苯环的特定构象相关:共面构象增加π-电子共轭程度,导致荧光红移,垂直构象减弱了其π-电子共轭效应,从而产生荧光蓝移。因此结合这个机制,最大发射波长发生蓝移的原因可能是加入水之后导致TPE的外围苯环结构偏向于垂直构象,从而导致化合物7最大发射波长发生蓝移[20]。
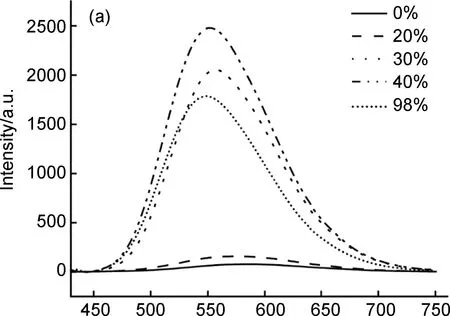
λ/nm
本文通过配位驱动自组装的方式合成了新型铂(II)金属大环化合物7,在未纯化下产率可达97%,几乎为定量合成。紫外可见吸收光谱表明:化合物7的吸收光谱几乎为化合物5和6的光谱叠加,结合核磁及质谱分析证明了目标产物的成功合成。荧光光谱测试表明:当混合溶液中水含量低于20%时,化合物7仅表现出微弱的荧光强度。随着水含量的进一步增加,荧光强度大幅增强,水含量为40%时达到最大值,约为纯DMSO中荧光强度的40倍,展现了化合物7优异的AIE性质,有望用于水环境中人工光捕获体系的构建。
猜你喜欢
上海化工(2018年10期)2018-10-31 01:21:06
机械工业标准化与质量(2018年1期)2018-03-21 05:09:41
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
科技资讯(2016年4期)2016-06-11 08:09:32
合成化学(2015年4期)2016-01-17 09:01:11
物理通报(2015年10期)2016-01-12 07:51:51
无机化学学报(2014年6期)2014-02-28 17:32:06
无机化学学报(2014年5期)2014-02-28 17:31:42
化工生产与技术(2014年5期)2014-02-27 13:42:02
钻探工程(2013年3期)2013-05-16 01:41:12
