
光/氧化还原酶协同催化的不对称合成研究进展
2024-04-27 09:40卓宇晴崔宝东
合成化学 2024年4期
卓宇晴, 崔宝东
(遵义医科大学 药学院,贵州 遵义 563003)
酶作为一种具有吸引力的用于不对称合成的催化剂,由于其专一性、高度选择性和反应条件温和等优点[1],越来越多地被用于不对称合成[2-4]。此外,由于酶活性口袋独特的空间结构可以降低反应活化能并调控反应过程,从而可以在温和条件下催化高度化学选择性、区域选择性和立体选择性的反应,在有机合成中能得到相对高纯度化学合成法无法得到的目标产物而备受青睐。然而,与化学催化相比,酶催化的反应类型仍相对较少。近十年来,光催化已发展成为广泛应用于有机合成的方法,其中可见光催化法拥有绿色清洁且条件温和的优点,并且可以用来构建一些独特的分子结构。光酶催化不仅能够利用光再生辅因子来发挥酶的天然活性,还能引发酶的非天然反应活性,获得新酶催化功能。另一方面,利用酶催化的高选择性和可定向进化的特性,使光酶催化能够调控光引发的活泼反应中间体,从而为光化学领域的立体化学控制难题提供新的解决方案。
氧化还原酶作为一类发展较早、研究较为成熟的生物酶,因在催化反应过程中常表现出极高的化学选择性、高催化效率和反应条件温和等优点,在手性药物和功能性高分子聚合物的不对称合成方面获得了广泛的应用。例如,氧化还原酶在催化羰基的不对称还原、碳氢键氧化、环氧化和拜耳维利格氧化等转化研究方面已发挥了重要作用,其中羰基的不对称还原是其应用于生物催化中最活跃的研究之一[5]。近年来,人们陆续发现氧化还原酶可以被光激发,从而诱导酶的混杂反应性。含有黄素和烟酰胺的酶如酮还原酶、烯烃还原酶等可以参与生物体中非光驱动的双电子氧化还原代谢过程,这些酶的辅因子可以通过与非天然底物形成电子供体-受体复合物(Electron donor-acceptor complex, EDA复合物),接着通过可见光激发诱发单电子转移(Single electron transfer, SET)生成自由基物种,实现光诱导的酶的非天然反应。EDA复合物产生于电子受体和供体之间的前线轨道的电子耦合,并可吸收可见光引发SET,随后生成自由基离子对,通过在底物中添加离去基团来促进自由基反应的进行。氧化还原酶利用一系列氢键作用来增强底物的亲电性,从而引发底物被各种辅因子还原,如果这种底物活化机制与单电子还原剂合并,就有可能使用离子机制的氧化还原酶通过自由基中间体进行反应[6-7]。通过光氧化还原催化实现酶促反应所需辅因子的再生,是光酶结合最直观的系统,也是最容易实现且报道最多的系统,其意义在于替代成本高昂的天然辅因子,或简化电子传递链从而实现更高效的反应。
研究者将这些生物系统的优点与人工化学催化反应相结合,设计出顺序、并发和协同性的化学酶催化过程[8-15](图1)。其中光与氧化还原酶协同催化是最常见的催化反应的结合方式,该反应过程通过光照的激发产生自由基,同时被酶的活性口袋所捕获进行后续一系列有趣的反应。光与氧化还原酶协同催化开辟了新酶催化合成应用研究的新领域,本文概述了近年来光与氧化还原酶协同催化在不对称合成研究领域的进展情况。

图1 顺序、并发和协同的化学酶催化过程
烯烃以热力学稳定的形式存在,通常反式的烯烃比顺式的烯烃稳定,因此烯烃往往是以反式结构参与各种反应过程。然而,在可见光催化下,反式烯烃能原位异构化为顺式烯烃,再由烯烃还原酶将顺式烯烃进行不对称还原反应,最终得到与常规反式烯烃相反构型的还原产物。该方法实现了烯烃异构体混合物的立体会聚式还原。
2018年,美国伊利诺伊大学香槟分校的赵惠民教授和加州大学伯克利分校的HARTWIG教授等[16]合作报道了一种从贝氏耶尔森氏菌(Yersinia bercovieri)中分离得到的烯还原酶和可见光协同催化的烯烃异构化/不对称还原反应。作者利用[Ir(dmppy)2(dtbbpy)]PF6或者黄素单核苷酸(FMN)作为光催化剂,将烯烃异构化,使其由反应活性较低的反式烯烃转化为不稳定的反应活性高的顺式烯烃,烯还原酶在辅因子再生的葡萄糖脱氢酶的存在下,专一性地将顺式异构体进行C=C的还原,由此可以高选择性地得到单一构型的对映异构体产物。结果表明,光催化剂和酶之间的相容性使化学酶过程能够超越辅因子再生(图2)。

图2 光酶协同催化的烯烃异构化/不对称还原反应
2020年赵惠民团队[17]通过利用光氧化还原再生黄素辅酶(FMNH2)的系统,在没有使用烟酰胺辅酶(NADPH)再生系统的情况下,以乙二胺四乙酸(EDTA)或三乙醇胺(TEOA)为终端还原剂,也实现了活化烯烃的协同立体汇聚式还原(图3)。
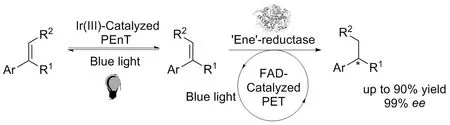
图3 光酶协同催化的活化烯烃的协同立体汇聚式还原
2018年,HYSTER课题组[18]利用烯烃还原酶与光氧化还原催化剂协同催化实现了α-乙酰氧基苯并环己酮的脱乙酰氧基反应(图4)。在530 nm可见光照射下,利用光催化剂Rose Bengal诱导烯烃还原酶活性口袋中通过氢键与底物结合单电子还原,脱去乙酸根阴离子后生成α-羰基碳自由基中间体,接着与烯烃还原酶口袋中的NADPH发生氢自由基转移(HAT),最终实现烯烃还原酶催化的对映选择性脱乙酰氧基反应。同位素标记实验证明,烟酰胺为氢原子的来源。另外,利用曙红Y为光催化剂,酮还原酶还可以催化α-溴酰胺的自由基脱卤化反应,该策略展示了光催化氧化还原与生物酶催化结合用于拓展天然酶应用的巨大潜力。

图4 光酶协同催化的α-乙酰氧基苯并环己酮的脱乙酰氧基反应
可见光与烯烃还原酶协同催化可用于活化烯烃、酮和烯酰胺的不对称还原反应。该协同催化策略不仅扩大了烯烃还原酶的底物应用范围,同时还揭示了一种区别于传统羰基不对称还原负氢转移的路径。
2019年,HYSTER课题组[19]证明了光与烯烃还原酶协同催化芳香酮类底物不对称还原的自由基历程(图5)。在反应过程中,烯烃还原酶首先与苯乙酮通过氢键作用降低其还原电位,使该氢键复合物在光催化的条件下通过选择性单电子还原生成酮基自由基中间体,该中间体再通过与黄素对苯二酚(FMNhq)作用完成HAT,实现烯烃还原酶对非天然酮类底物的对映选择性还原。实验机理表明,该反应通过酮基自由基的形成而进行,且不同于传统的负氢转移还原羰基的反应路径。此外,该反应可以在未对酶或辅因子进行任何修饰的情况下进行,从而允许原生和非天然的机制同时发生。

图5 光酶协同催化的芳香酮的不对称还原反应
2020年,HYSTER课题组[20]实现了烯烃还原酶与可见光协同催化的乙烯基吡啶类底物的不对称还原反应。实验机理表明,该反应通过自由基机制进行,其中乙烯基吡啶在溶液中得到1个单电子被还原成相应的苄基自由基负离子。通过离散傅里叶变换(DFT)计算显示,自由基是“动态稳定的”,表明自由基具有足够长的寿命,可以与酶的活性位点相结合进行立体选择性氢原子转移,该反应也进一步证明了光酶协同催化对于开发天然酶新功能的重要(图6)。

图6 光酶协同催化的乙烯基吡啶的不对称还原反应
2021年,HYSTER课题组[21]证明了通过可见光的激发,烯烃还原酶可以由单电子转移机制实现丙烯酰胺类底物的不对称还原。研究机理表明,自由基的形成是通过FMNhq和蛋白质活性位点内的缺电子底物之间形成的三元电荷转移(CT)复合物的光激发而完成的,只有当2种底物都存在于蛋白质活性位点时才会发生自由基的形成。因此,2个偶联体的空间取向对CT复合物的形成至关重要(图7)。

图7 光酶协同催化的丙烯酰胺的不对称还原反应
2023年,黄小强课题组[22]报道了一种390~430 nm的可见光与烯烃还原酶ER1的协同催化策略。通过可见光的激发,烯烃还原酶ER的反应活性首次扩展到烯酰胺的单电子还原,从而不对称构建了手性胺(对映体比例高达99 ∶1)。该方法是对传统过渡金属催化烯胺不对称还原方法的重要补充。研究机理表明,对苯二酚态黄素辅因子的直接可见光激发可以有效增强烯烃还原酶的还原能力,从而触发生物催化单电子的还原过程。温和、绿色的反应条件以及广泛的官能团耐受性使这种非自然的光酶催化策略成为传统化学催化方法的重要补充(图8)。
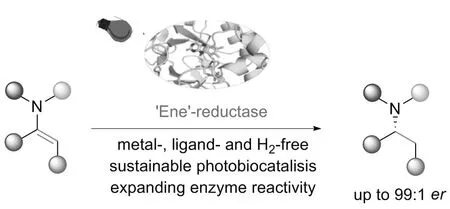
图8 光酶协同催化的烯酰胺的不对称还原反应
2020年,HYSTER课题组[23]报道了一种光酶协同催化氧化还原反应的策略,通过手性β-取代酮原位消旋化和不对称羰基还原协同兼容的反应过程,实现了β-取代的手性醇的合成。在整个反应过程中,首先β-取代酮底物与二级胺作用脱水生成烯胺,烯胺在以Ir(ppy)2(dtb-bpy)PF6为光敏剂和n-OctSH为还原剂的白光LED照射下,经单电子转移生成β-碳自由基烯胺中间体,该中间体进一步在酶的催化下经不对称氢转移和烯胺水解产生β-取代的羰基自由基,经进一步不对称羰基还原生成最终的β-取代的手性醇。该机理表明,烯胺中间体是经过光照激发下单电子氧化得到的,这使羰基β-位的C—H键酸性增强,从而促进去质子化过程,有利于使羰基β-位静态立体中心发生外消旋化(图9)。
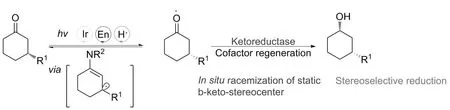
图9 光酶协同催化的β-取代酮原位消旋化/不对称还原反应
分子内的不对称氧化还原偶联反应可以用于构建不同环大小的化合物。通过可见光与烯烃还原酶协同催化分子内的不对称脱卤或脱羧C—C键偶联反应可以高效构建手性内酰胺、环烷烃以及苯并含氧杂环等化合物,进一步扩展了光酶协同催化不对称合成的反应类型。
2019年,HYSTER课题组[24-25]报道了光和烯烃还原酶协同催化的分子内自由基环化反应,构建了几类对映体富集的五元、六元、七元和八元内酰胺类化合物(图10)。研究结果表明,黄素依赖型“烯”还原酶(EREDs)通过单电子转移机制催化具有高立体选择性的自由基反应,该途径不同于传统的氢化物转移的机理路径,缺电子的α-氯代酰胺与ERED活性位点内的FMNhq可以形成CT复合物。当复合物被可见光照射时,α-氯代酰胺底物经脱卤并从黄素中得到1个电子,从而产生亲电性的自由基。整个过程的立体化学控制是通过酶口袋对自由基加成方向以及后续的HAT步骤限制实现的。

图10 光酶协同催化α-氯代酰胺的分子内自由基环化反应
2019年,HYSTER课题组[26]证明黄素依赖性“烯”还原酶(ERED)OPR1与可见光协同催化可以实现α-氯代酰胺的不对称分子内自由基环化反应。该策略以α-氯代酰胺为起始底物,可以对映选择性合成含3,3-二取代氧化吲哚骨架结构的化合物。研究机理表明,该反应在黄素半醌/醌氧化还原对存在的条件下进行,其中电子从FMNhq转移到基态黄素半醌,黄素半醌作为还原剂提供电子,黄素醌氧化环化后形成乙烯基α-酰胺自由基,最后黄素半醌(FMNsq)完成氢原子转移。这种机理对于该酶家族以前是未报道的,突出了ERED在不对称合成中的多功能性(图11)。

图11 光酶协同催化的α-氯代酰胺的分子内自由基环化反应
2020年,CLAYMAN课题组[27]报道了一种光与烯烃还原酶协同催化的以烷基碘化物作为不稳定亲电自由基前体参与的对映选择性自由基环化反应。研究机理表明,这种自由基反应过程是通过光激发蛋白质活性位点内黄素和底物之间形成的电荷转移复合物实现的。缺电子的α-氯代酰胺与ERED活性位点内的FMNhq形成CT复合物,当这种复合物被可见光照射时,底物脱卤并从黄素得到1个电子,产生亲电性的碳自由基。接着,该自由基对不饱和双键进行分子内的立体选择性加成反应,形成C—C键并产生新的碳自由基,该自由基最后从黄素半醌得到氢自由基,生成最终的手性环化产物(图12)。
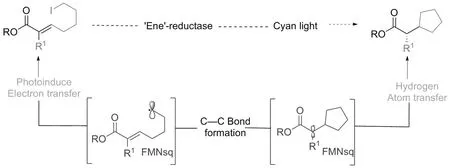
图12 光酶协同催化的烷基碘化物的对映选择性自由基环化反应
2022年,HYSTER课题组[28]报道了利用黄素依赖性“烯”还原酶和外源性蓝光催化剂协同催化的不对称分子内自由基反应,实现了手性六元环内酰胺类化合物的不对称构建。该方法以不饱和的酰基羟胺为底物,在烯还原酶YqjM和光催化剂Ru(bpy)3Cl2存在下,通过蓝光激发和定向进化技术,找到了1个三突变体YqjM-R(M25L-C26S-I69T),可以91%的产率、97 ∶3的er(对映体比例)得到R构型的目标产物。基于YqjM的Y28位点,经过5轮的进化,成功找到了1个五突变体YqjM-S(Y28A-I69N-Y169H-A252G-R336W),可以89%的产率、14 ∶86的er得到S构型的目标产物。这些酶通过定向进化进行工程设计,可以催化5-外、6-内、7-内、8-内和分子间的对映选择性氢胺化反应。研究机理表明,自由基的引发是通过酶的门控调节机制发生的。其中,蛋白质可以通过光催化剂热力学激活底物的还原完成电子转移。该方法证明了这些酶在控制高能自由基中间体反应性方面的多功能性,通过光酶协同催化策略实现了之前酶催化无法单独实现的功能(图13)。

图13 光酶协同催化不对称分子内自由基反应用于构建酰胺化合物


图14 光酶协同催化用于不对称合成苯并氧杂环化合物
除光酶协同催化的分子内自由基环化反应用于不对称构建手性环状化合物外,近年来,光酶协同催化逐步拓展到分子间的不对称C—C键偶联反应,进一步拓宽了其合成应用范围。通过光与烯烃还原酶协同催化α-卤代烃、苯磺酰氯、亚磺酸钠盐与烯烃和硝基烷烃的不对称氧化还原C—C键偶联反应以及N-芳基α-氨基酸与乙烯基吡啶的不对称脱羧偶联反应,不断丰富和拓展了光酶协同催化在不对称构建C—C键的合成应用。
2020年,赵惠民课题组[30]报道了可见光诱导的烯还原酶催化的非天然两分子交叉偶联反应。通过烯还原酶/光协同催化策略,实现了化学合成方法难以得到的含γ-手性碳中心的手性酮类化合物。从烯烃和α-卤代羰基化合物出发,通过可见光诱导烯烃还原酶催化的自由基氢烷基化途径的分子间偶联反应,实现γ-手性羰基化合物的高效构建,反应过程具有优异的转化率和对映选择性。机理研究表明,α-卤代羰基化合物通过氢键作用结合在酶的位点上,并和半醌型黄素单核苷酸(FMNH)作用生成电子供体-受体EDA复合物。这种EDA复合物受光激发完成单电子转移过程,同时脱除羰基α-碳上的卤原子,生成FMNH和α-羰基自由基。随后,α-羰基自由基对烯烃分子进行加成反应,并在烯还原酶的立体控制作用下从FMNH获取氢自由基完成反应,最终生成黄素单核苷酸(FMN)和手性产物(图15)。
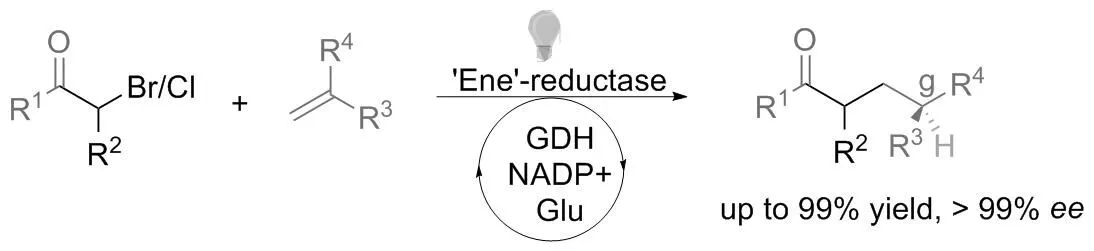
图15 光酶协同催化α-卤代羰基化合物与烯烃的交叉偶联反应
2022年,HYSTER课题组[31]还实现了光酶协同催化的α-卤代酮和硝基烷烃的不对称Csp3—Csp3亲电交叉偶联反应(图16)。在酶辅因子作用下,α-卤代酮能够与酶作用形成电荷转移复合物,α-卤代酮通过光照激发能够被化学选择性还原产生烷基自由基。在C—C键形成的过程中,烷基自由基与原位产生的硝酸盐反应形成硝基自由基阴离子,然后该阴离子分解形成亚硝酸盐和烷基自由基,最后经酶控制的氢转移完成整个自由基反应过程,同时实现高水平的对映选择性控制。HYSTER课题组筛选出2种烯烃还原酶CsER和GluER-T36A,可分别催化获得S-对映体和R-对映体富集产物的生成。值得注意地是,与之前常用的偏中性的缓冲体系不同,pH=9.0的反应缓冲液有利于硝基烷烃的去质子化。这种亲电性反应在小分子催化中是难以实现的,突出了酶催化新机制用于解决长期存在的合成挑战的潜力。

图16 光酶协同催化的不对称Csp3-Csp3亲电交叉偶联反应
2022年,HYSTER课题组[32]首次报道了由黄素依赖性“烯”还原酶催化的α-氯代酰胺与烯烃分子间的不对称C—C键偶联反应。在反应过程中,有机卤化物通过与FMNhq在“烯”-还原酶(EREDs)的活性位点内形成CT复合物,参与各种分子间的C—C键形成反应,通常具有较高ee值。研究机理表明,自由基是通过烯烃、α-氯代酰胺和黄素对苯二酚之间形成的罕见的高阶酶模板电荷转移复合物的光照激发而产生。这种独特的机制说明只有当2种底物都存在于蛋白质活性位点内时,才会形成自由基。同时该活性位点可以有效控制碳自由基与氢自由基的立体转移结合过程,从而确保得到高选择性的γ-手性酰胺类产物。这项工作进一步激发了光酶协同催化的应用潜力,可以通过以前未知的电子转移机制实现新的生物催化转化(图17)。

图17 光酶协同催化α-氯代酰胺与烯烃的不对称分子间C—C键偶联反应
2023年,HYSTER课题组[33]将传统的烯烃类化合物拓展到了吲哚杂环化合物,实现了烯烃还原酶(ERED)(GluER-T36A)与390 nm可见光协同催化的杂芳烃的区域选择性烷基化反应。通过酶的定向进化改造,可催化选择性获得吲哚C4-位烷基化的产物。机理研究表明,蛋白质活性位点的变化会改变负责自由基形成的CT复合物的电子特性,导致在CT复合体中产生基态CT的变体。这项研究突出了酶可以高区域选择性调控自由基反应,而传统的小分子催化剂很难实现这种反应的区域选择性调控(图18)。

图18 光酶协同催化杂芳烃的区域选择性烷基化反应
2023年,HYSTER课题组[34]提出了一种利用黄素依赖性“烯”还原酶与390 nm的紫光进行协同催化的策略,突破性实现了非活泼氯甲基吡啶杂环作为烷基自由基前体与烯烃的不对称C—C键交叉偶联反应,构建了一类含γ-手性碳中心的吡啶类化合物。反应从简单易得的氯甲基吡啶与缺电子的杂环化合物出发,通过与端位烯烃的C—C键偶联反应有效增加简单杂环结构的复杂性。研究机理表明,这种光驱动的转化是通过瞬时酶-底物复合物的激发启动,使酶能够在底物结合活性位点时产生自由基,从而引发所需的还原电位。这项工作是一种立体选择性合成吡啶和类似杂环衍生物的代表性方法,扩展了化学合成中“烯”还原酶的底物适用范围(图19)。

图19 光酶协同催化用于构建含γ-手性中心的吡啶类化合物
2023年,HYSTER课题组[35]进一步拓展了光酶协同催化的合成应用范围,发展了一种黄素依赖性的烯还原酶老黄酶与蓝光协同催化N-芳基甘氨酸与乙烯基吡啶的氧化还原中性脱羧偶联反应,以高达92%的产率和99 ∶1的er获得脱羧偶联产物。此外,该课题组运用立体互补的酶作为生物催化剂,可以催化获得R和S2种不同构型的对映体产物。机理研究表明,外源钌光催化剂Ru(bpy)32+辅因子与蛋白质结合,有助于在酶的活性位点定点形成自由基(图20)。

图20 光酶协同催化乙烯基吡啶的氧化还原中性脱羧偶联反应
2023年,徐鉴课题组[36]运用光酶协同的催化策略,首次将传统的烷基自由基拓展到磺酰基自由基,实现了自由基介导的烯烃的立体选择性氢磺酰化反应。通过对烯烃还原酶进行定向进化,构建了老黄酶OYE1的变体Y82F,实现了4-甲基苯磺酰氯和α-甲基苯乙烯的立体选择性氢磺酰化反应(对映体比例高达99 ∶1)。并考察了该反应的底物应用范围。最后通过紫外可见光谱、氘代实验和分子动力学模拟对反应的机制进行了解析。该研究为手性砜类化合物的合成提供了一类新方法,并进一步扩展了黄素依赖酶的催化合成应用范围(图21)。

图21 光酶协同催化的烯烃的立体选择性氢磺酰化反应
2024年,叶俊涛课题组[37]报道了一例全新的光酶协同催化的单电子氧化引发的烯烃的对映选择性氢磺酰化反应。该课题组利用廉价易得的芳基或烷基亚磺酸钠盐为自由基前体,在不依赖底物和辅因子的条件下,通过与酶形成电子供体-受体复合物,成功实现了非环状烯烃的对映选择性氢磺酰化反应,高产率和高对映选择性合成了一系列手性砜类化合物。更重要地是,GluER变体具有高催化活性和对映选择性以及广泛的底物适用。机理研究表明,实验支持提出的单电子氧化引发的机理,并揭示了酪氨酸残基Y343可能是不对称HAT步骤的最终氢原子给体。考虑到易氧化的自由基前体种类繁多且酶工程技术的日趋成熟,单电子氧化引发的光酶催化体系有望在未来实现更多类型和更具挑战性的合成转化(图22)。

图22 光酶协同催化烯烃的对映选择性氢磺酰化反应
光酶催化反应尽管才刚刚兴起,但是该策略已经在合成科学领域展现出巨大的应用前景。总体来说,光酶协同催化是把酶催化的生物优势和光催化的化学优势结合到一起,填补了目前现有生物催化和化学催化领域的一些空白。将生物催化与光催化相结合,光催化能产生高活性的反应中间体,并结合生物催化在控制立体选择性方面的独特优势,通过利用光子吸收的能量驱动化学转化,是实现更加绿色化学的有力策略,同时也能很好地利用催化中辅因子的循环,达到节约成本或者更高效电子传递等目的。但是,光酶协同催化反应所用的酶主要集中于烯烃还原酶、酮还原酶等氧化还原酶类,协同催化的酶类比较单一,并且协同催化的反应类型还比较有限,主要集中于光/氧化还原酶协同催化的不对称还原反应以及分子内/分子间的不对称氧化还原偶联反应等。随着新酶、酶新催化功能和光酶协同催化机制的不断发现,未来将进一步挖掘光酶协同催化在有机合成中的应用潜力,并不断开发出更多的可见光与其他酶类协同催化的有趣的反应类型。
猜你喜欢
云南化工(2021年8期)2021-12-21
昆明医科大学学报(2021年5期)2021-07-22
中国石油石化(2021年9期)2021-07-17
海峡姐妹(2019年8期)2019-09-03
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
当代化工研究(2016年2期)2016-03-20
应用化工(2014年12期)2014-08-16
中国酿造(2014年9期)2014-03-11
