
TYK2抑制剂BMS986202的合成工艺
2024-04-27 09:40:06张倩倩李铭东党诗涵李庶心胡文祥
合成化学 2024年4期
张倩倩, 李铭东, 党诗涵,, 李 春,,, 李庶心, 胡文祥,4*, 闵 清*
(1. 湖北科技学院 药学院,湖北 咸宁 437100; 2. 江西中医药大学 药学院,江西 南昌 330000; 3. 苏州隆博泰药业有限公司,江苏 苏州 215000; 4. 北京神剑天军医学科学研究院 京东祥鹄微波化学联合实验室,北京 101601)
Deucravacitinib(BMS986165)是全球首款上市的口服选择性酪氨酸激酶2(TYK2)变构抑制剂,也是首款原创性氘代新药。BMS986165由百时美施贵宝(BMS)公司研发,化学结构见图1,并于2022年9月9日被美国食品和药物管理局批准用于适合全身治疗或光疗的中重度斑块型银屑病成人患者[1]。 TYK2也是JAK家族,包括JAK1, JAK2, JAK3和TYK2中的一员。JAK抑制剂在炎症和癌症等治疗领域取得了很大成功,但它们在临床应用中容易引起严重感染、恶性肿瘤和血栓等严重副作用[2-4]。由于调控的细胞信号通路不同,相比于副作用严重的JAK抑制剂,高选择性的TYK2在治疗多种自身免疫性疾病,例如斑块型银屑病[5]、银屑病关节炎[6]、系统性红斑狼疮[7]、炎性肠病[8]、类风湿性关节炎[9]和斑秃[10]等的同时,避免了泛抑制带来的副作用[11]。 BMS986165选择性结合TYK2假激酶(JH2)结构域,并通过稳定调节JH2结构域来阻断受体介导的TYK2激活,从而抑制白细胞介素IL-23、 IL-12和干扰素IFN的信号传导[12]。 BMS986165选择性与TYK2的调控域结合,实现对TYK2及其下游信号的变构抑制,相对其他JAK亚型具有较高的选择性,在建议的治疗剂量下,不会抑制JAK1, JAK2和JAK3的活性[13]。

图1 BMS986165的结构
BMS986202(图2)是百时美施贵宝公司基于BMS986165发现的一种新的临床候选药物。研究人员基于结构优化,将BMS986165骨架中的哒嗪优化为吡啶,再对苯环取代基杂环的对位进行取代,以占据朝向Thr599残基的疏水空腔,由此发现了BMS986202。 BMS986202具有较好的活性、水溶性和渗透性。经过进一步的药代性质评价,研究人员发现其具有较优秀的药动学性质,口服生物利用度极好。同时,研究人员通过IL-12/IL-18诱导小鼠干扰素产生的体内模型、IL-23诱导的皮肤炎症模型和结肠炎模型,系统评价了化合物BMS986202的体内活性,发现其均表现出显著的体内活性[14]。目前,BMS986202正处于Ⅱ期临床试验,具有良好的开发和应用前景。

图2 BMS986202的结构
目前,专利[15]报道的BMS986202合成方法主要是以2-溴-6-硝基苯酚为起始原料,经取代、还原、偶联和芳基化,最后再与环丙甲酰胺进行酰胺化反应制得。步骤较少,收率较高,但在中间体的纯化上,需要通过柱层析来完成,存在反应周期长,负载量低,时间成本增加的问题,不适合工业化生产。
本研究参考原研路线(图3),对BMS986202的合成工艺进行了优化,产品结构经1H NMR分析表征。制备中间体2时,原研专利[15]所用溶剂为乙醇和水,反应温度为室温,通过柱色谱纯化,收率为85%。本研究以甲醇作为溶剂,控制反应温度约20 ℃,反应达到完全且杂质少,无需纯化可直接用于后续反应,避免了原研专利通过快速柱色谱纯化产物,大幅度地简化了操作,最终收率为81%。制备中间体4时,原研专利[15]的合成路线以6-溴-2-硝基苯酚为起始原料,经取代、还原和偶联3步反应制得(图4),收率为60%。然而3步反应步骤繁琐,中间体均需要通过柱层析进行纯化,存在操作复杂、耗时长和成本增加的问题,不适合放大生产。因此,本研究直接将1-溴-2-甲氧基-3-硝基苯作为原料,既简化了步骤,又节省了时间和成本。起初,以1-溴-2-甲氧基-3-硝基苯为原料,想先通过偶联反应再还原制得中间体4(图5)。然而,在实验过程中,本研究发现大约有75%的原料未反应完全,收率很低。因此,最终选择先还原再偶联的方法,最后收率为72%。在制备中间体6时,原研专利[15]采用的钯试剂为[1,1′-二(二叔丁基膦)二茂铁]合二氯钯(II),碱为磷酸氢二钾,反应温度为100 ℃,收率较低,为75%。随后,本实验将钯试剂改为1,1′-双二苯基膦二茂铁二氯化钯,碱改为碳酸钠,反应温度降至75 ℃,体系干净,杂质少,后处理简单,收率得到提高,为81%。制备中间体7时,将氯代试剂改为氯化亚砜,碱改为三乙胺,反应时间缩短,纯度提高,后处理简单,收率由原研专利[15]的74%提高到81%。而重复专利[15]制得中间体7,反应时间长,杂质多,需要通过柱层析进行纯化,操作复杂且繁琐。制备中间体8时,本研究使用四氢呋喃为反应溶剂,沸点低,避免专利[15]中处理N,N-二甲基乙酰胺的步骤,后处理更加简单,方便,收率由79%提高至84%。制备目标化合物BMS986202时,本研究将原研专利[15]的溶剂由二氧六环改为1,4-二氧六环和水的混合溶剂,反应温度由130 ℃降至90 ℃,反应收率由原研专利的70%提高到85%。

图3 BMS986202的合成路线[15]

图4 中间体4的合成路线[15]

图5 本文中间体4的合成路线
1 实验部分
1.1 仪器与试剂
ZF-20C型暗箱式自动紫外分析仪; RY-1G型熔点仪(温度未校正); Varian 400 Hz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标)。
化合物1,成都伊诺达博医药科技有限公司;化合物3,毕德医药有限公司;化合物5,江苏艾康生物医药研发有限公司;化合物9,萨恩化学技术(上海)有限公司;甲醇、叔丁醇、1,4-二氧六环,江苏强盛功能化学有限公司;四氢呋喃,无锡市晶科化工有限公司。所用试剂均为分析纯。
1.2 合成
(1) 3-溴-2-甲氧基苯胺(中间体2)的合成
将原料1-溴-2-甲氧基-3-硝基苯(30.0 g, 129.3 mmol)加入甲醇溶液(80 mL)中,再加入氯化铵(45.0 g, 841.3 mmol)。将其置于冰水浴中搅拌10 min,然后缓慢加入锌粉(67.2 g, 103.4 mmol),速度以控制体系温度不超过20 ℃为宜,加毕,继续冰水浴下(0 ℃)反应2 h(TLC检测)。体系过硅藻土抽滤,滤液减压浓缩,加入饱和碳酸钠水溶液(150 mL),然后用乙酸乙酯(3×50 mL)萃取,合并有机相,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,得到粗品中间体221.2g,收率81%,直接投入下一步反应。
(2) 2-甲氨基-3-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯胺(中间体4)的合成
将原料3-溴-2-甲氧基苯胺(21.2 g, 90.6 mmol)和联硼酸频那醇酯(27.6 g, 108.7 mmol)依次加入叔丁醇(80 mL)溶液中(20 ℃),电磁搅拌,然后加入2-乙基己酸钾(49.5 g, 271.7 mmol),抽真空,N2置换,加入PdCl2(dppf)(0.7 g, 0.9 mmol),再次抽真空,N2置换3次,然后加热,升温至80 ℃回流反应4 h(TLC检测)。体系过硅藻土抽滤,滤液减压浓缩后加入冰水(150 mL),然后用乙酸乙酯(3×50mL)萃取,合并有机相,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经石油醚打浆纯化得中间体4,棕色固体,16.3 g, m.p.101~103 ℃,收率72%;1H NMR(400 MHz, CDCl3)δ:7.14~7.11(m, 1H), 6.93(t,J=15.2 Hz, 7.6 Hz, 1H), 6.87~6.85(m, 1H), 3.83(3, 3H), 1.36(s, 12H)。
(3) 3-(5-氟嘧啶-2-基)-2-甲氧基苯胺(中间体6)的合成
将碳酸钠(17.4 g, 163.8 mmol)加入250 mL三口烧瓶中,加水(60 mL)搅拌,然后加入原料2-甲氨基-3-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯胺(16.3 g, 65.5 mmol)和2-溴-5-氟嘧啶(12.7 g, 72.1 mmol),再滴加1,4-二氧六环(70 mL),电磁搅拌,抽真空,N2置换,加入PdCl2(dppf)(2.7 g, 3.7 mmol),再次抽真空,N2置换3次,加热,升温至75 ℃反应4 h(TLC检测)。体系过硅藻土抽滤,滤液减压浓缩,加入冰水(150 mL),用乙酸乙酯(3×50mL)萃取,合并有机相,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,得中间体6,黄色油状液体,11.6 g,收率81%,粗品,直接投入下一步反应。1H NMR(400 MHz, DMSO-d6)δ:8.96~8.94(d,J=0.8 Hz, 2H), 7.23~7.20(m, 1H), 7.15~7.12(m, 1H), 6.95~6.90(m, 1H), 5.25(s, 2H), 3.83(s, 3H)。
(4) 4,6-二氯吡啶-氘甲基酰胺(中间体7)的合成
将原料4,6-二氯烟酸(20.0 g, 104.2 mmol)投入250 mL三口烧瓶中,然后加入氯化亚砜(50 mL),同时滴加催化剂量的N,N-二甲基甲酰胺,电磁搅拌。加热,升温至80 ℃回流反应。体系逐渐澄清、透明,呈淡黄色。1.5 h后(TLC检测),将体系浓缩,旋干。将残留物溶于二氯甲烷中,再次浓缩,旋干,以确保完全除去多余的二氯亚砜,得到化合物4,6-二氯烟酰氯,直接用于下一步反应。提前配置游离的氘代甲胺溶液:氘代甲氨盐酸盐(8.1 g, 114.6 mmol)加水,再加入三乙胺(11.6 g, 114.6 mmol),同时滴加二氯甲烷溶液(20 mL)。配置好后为透明混悬液,封口放入冰箱冷藏。将化合物4,6-二氯烟酰氯溶于二氯甲烷溶液(30 mL)中,然后将其置冰水浴冷却至0 ℃,缓慢滴加配置好的氘代甲胺溶液,滴加速度以保持体系温度不超过5 ℃为宜。滴加过程中,体系中有白色固体析出。滴加完毕,继续冰水浴下反应2.5 h(TLC检测)。加冰水(150 mL)将体系淬灭,用二氯甲烷(3×50 mL)萃取,合并有机相,用饱和K2CO3水溶液(50 mL)洗涤,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经石油醚打浆,纯化,得到中间体7,白色固体,17.5 g,收率81%, m.p.135~137 ℃;1H NMR(400 MHz, DMSO-d6)δ:8.57(s, 1H), 8.45(s, 1H), 7.90(s, 1H)。
(5) 6-氯-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基烟酰胺(中间体8)的合成
将3-(5-氟嘧啶-2-基)-2-甲氧基苯胺(11.6 g, 58.2 mmol)和中间体7(12.1 g, 57.9 mmol)依次加入装有四氢呋喃(30 mL)的250 mL三口烧瓶中,电磁搅拌;将其置于冰水浴中,待温度降至0 ℃时,缓慢滴加LiHMDS(15 mL),滴加速度以控制体系温度不超过20 ℃为宜,滴加完毕,撤去冰水浴,体系自然升至室温(21 ℃)反应3 h(TLC检测)。体系由墨绿色逐渐变成棕色。体系加冰水(5 mL)淬灭,过硅藻土抽滤,滤液减压浓缩,蒸除体系中的四氢呋喃,然后加入冰水(150 mL),用二氯甲烷(3×50 mL)萃取,合并有机相,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经打浆(二氯甲烷 ∶石油醚=5 ∶1,V∶V)纯化,过滤,收集滤饼,得中间体8,白色固体,18.2 g,收率84%, m.p.193~195 ℃;1H NMR(400 MHz, DMSO-d6)δ:10.31(s, 1H), 9.15(s,1H), 9.06~9.04(d,J=0.9 Hz, 2H), 7.95~7.90(s, 1H), 7.82~7.78(s, 1H), 7.77~7.75(m, 1H), 7.60~7.56(m, 1H), 7.46~7.41(m, 1H), 3.70(s, 3H)。
(6) 6-环丙烷甲酰胺基-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基烟酰胺的(BMS986202)合成
将碳酸铯(20.8 g, 63.8 mmol)投入250 mL三口烧瓶中,加水(30 mL)搅拌,然后加入原料6-氯-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基烟酰胺(10.0 g, 25.6 mmol),并滴加1,4-二氧六环(50 mL),抽真空,N2置换,加入Pd2(dba)3(1.2 g, 1.3 mmol)和Xantphos(1.5 g, 2.6 mmol),再次抽真空,N2置换3次,加热,升温至90 ℃反应4.5 h(TLC检测)。体系过硅藻土抽滤,滤液减压浓缩,蒸除反应溶剂后加入冰水(150 mL),用二氯甲烷(3×50 mL)萃取,合并有机相,再用饱和氯化钠水溶液(50 mL)洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经重结晶(二氯甲烷∶石油醚=3∶1,V∶V)纯化,得产物BMS986202,白色固体,9.5 g,收率85%,纯度98.8%, m.p.250~252 ℃;1H NMR(400 MHz, DMSO-d6)δ:10.73(s, 1H), 10.56(s, 1H), 9.05(d,J=0.9 Hz, 1H), 8.57(s, 1H), 8.52 (s, 1H), 8.14~7.95(m, 1H), 7.55(dd,J=7.8 Hz, 1.5 Hz, 1H), 7.43(dd,J=7.6 Hz, 1.3 Hz, 1H), 7.34~7.20(m, 1H), 3.65 (s, 3H), 2.08~1.84(m, 1H), 0.82~0.75(m, 4H); MS(ESI)m/z:calcd for C22H18D3FN6O3{[M+H]+}439.18, found 439.5。
2 结果与讨论
2.1 中间体4的合成分析
在合成中间体4时,主要考察了钯试剂、配体、碱和溶剂对Suzuki偶联反应的影响。结果发现,该类反应在大多数情况下,原料未能反应完全。因此本研究尝试了Pd2(dba)3和PdCl2(dppf) 2种钯试剂,实验结果显示,使用Pd2(dba)3和配体Xantphos时,体系未反应。本研究采用了无需配体参与的钯试剂PdCl2(dppf),发现原料发生反应。同时在以PdCl2(dppf)为钯试剂的前提下,对该反应的溶剂和碱进行了考察,最终确定了在反应温度为80 ℃条件下,以叔丁醇为溶剂,以2-乙基己酸钾为碱是合成中间体4的最佳条件,收率为72%(表1, Entry 5)。
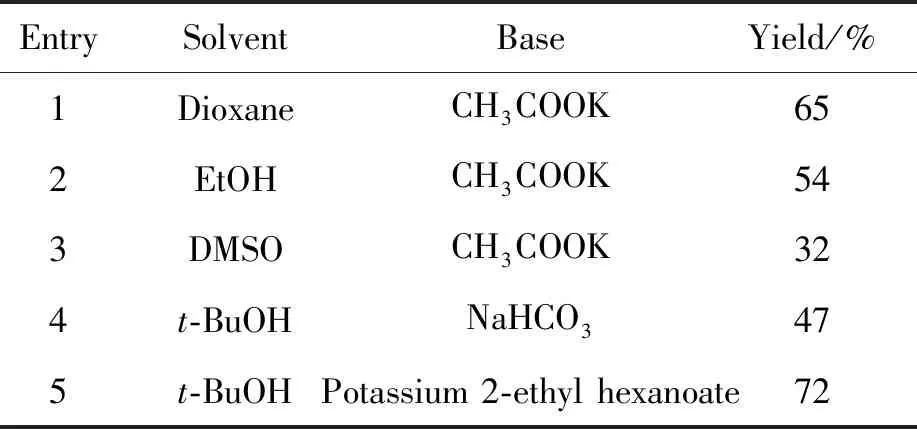
表1 溶剂、碱对中间体4收率的影响
2.2 中间体6的合成分析
类似中间体4,中间体6的合成过程同样考察了钯试剂、配体、碱、溶剂和温度对Suzuki偶联反应的影响。
(1) 碱对Suzuki偶联反应的影响
从Suzuki偶联反应的机理可以看出,碱在整个反应过程中起着重要的作用[16]。通常用于Suzuki偶联反应的碱既有有机碱也有无机碱。常用的无机碱有K3PO4、 K2CO3、 KOH、 Cs2CO3、 Na2CO3、 KF、 CsF和Ba(OH)2等;常用的有机碱有KOBu-t、 NaOBu-t、 KOMe、 NEt3和t-BuNH2等。本研究尝试了Na2CO3, K3PO4和K2HPO43种碱对Suzuki偶联反应的影响,实验结果显示,K3PO4和K2HPO4均不能使原料反应完全,收率较低。而用Na2CO3作为碱进行反应时,原料反应完全,体系杂质少,后处理简单,因此最终采用Na2CO3作为碱进行反应(表2, Entries 1, 4)。
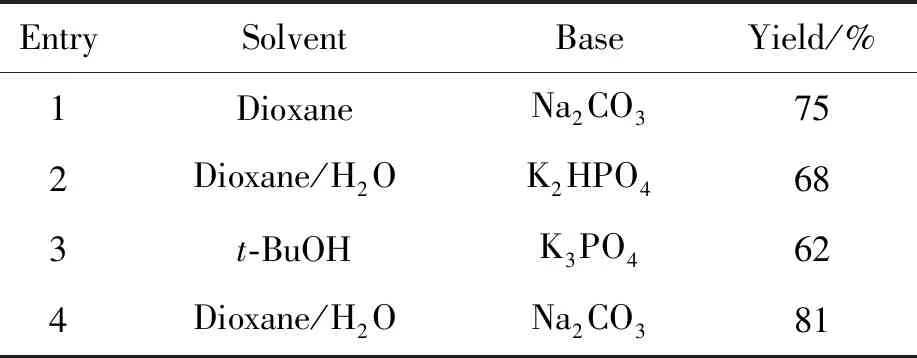
表2 溶剂、碱对中间体6收率的影响
(2) 反应溶剂对Suzuki偶联反应的影响
同碱一样,溶剂在Suzuki偶联反应中也起着重要的作用。它除了可以使参与反应的各个组分处于均相之外,也是调节反应温度的载体。在Suzuki偶联反应中各类溶剂均有应用,例如:DMF、二氧六环、THF、甲苯、二甲苯、乙腈、三氯甲烷、丙酮以及各种醇等。醇中包括甲醇、丙醇和丁醇等。此外,一些混合溶剂在该反应中也有很好的表现。本研究尝试了1,4-二氧六环、叔丁醇以及混合溶剂1,4-二氧六环/水3种溶剂对Suzuki偶联反应的影响,结果发现,当反应溶剂为1,4-二氧六环 ∶水=4 ∶1(体积比)时,原料反应完全,体系干净,收率较高,为81%(表2, Entry 4)。
(3) 反应温度对Suzuki偶联反应的影响
在本实验过程中发现,不同的反应温度对化合物的合成也有一定影响。专利报道的温度为100 ℃。因此,进一步考察不同反应温度对中间体6收率的影响,结果显示,温度超过100 ℃时,反应体系杂质多,原料大部分未反应完全;温度为80~100 ℃时,反应体系杂质减少,原料大部分反应完全;温度为70~80 ℃时,反应体系干净,原料几乎反应完全,产物纯化简单,收率高,因此,选择反应温度为70~80 ℃,具体结果见表3。

表3 反应温度对中间体6收率的影响
综上所述,在该反应中,选择反应溶剂为1,4-二氧六环/水,碳酸钾为碱,反应温度为70~80 ℃,中间体6收率可达到81%。
2.3 中间体7的合成分析
本研究对已有的合成路线进行了优化,在合成中间体7时,将羧酸制成酰氯,再与氘代甲胺进行酰胺化反应。本研究首先尝试了原研专利的合成方法,并对其进行了优化,尝试了2种氯代试剂及碱对酰化反应的影响。实验结果显示,采用原研专利的合成方法,以草酰氯为氯代试剂,N,N-二异丙基乙胺为碱时,反应时间较长,反应体系较杂,需要柱层析进行纯化,收率低;使用氯化亚砜(SOCl2)为氯代试剂,三乙胺(TEA)为碱时,反应快,杂质少,只需要通过打浆对产物进行纯化。因此,本文确定以SOCl2为氯代试剂,TEA为碱,收率为81%(表4, Entry 2)。
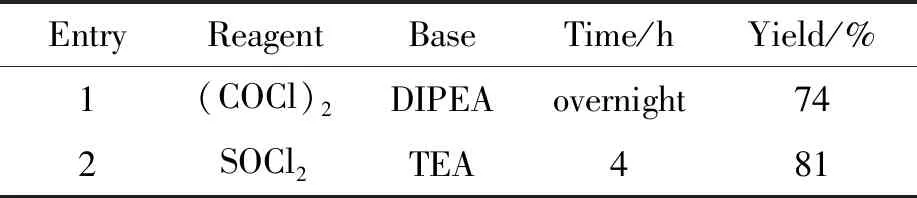
表4 氯代试剂及碱对中间体收率的影响
本文以1-溴-2-甲氧基-3-硝基苯为起始原料,通过还原、偶联、芳基化和酰胺化4步反应,确立了有效合成BMS986202的反应路线,并对反应路线中的关键工艺条件进行了优化。通过1H NMR, MS(ESI)对目标产物BMS986202进行了表征,总收率达85%。合成BMS986202的工艺路线稳定,反应过程温和,操作简便且收率较高,可为进一步进行工业化生产打下良好基础。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
药品评价(2020年13期)2020-10-14 03:50:40
工业设计(2020年6期)2020-07-30 14:05:39
中国生殖健康(2019年2期)2019-08-23 08:11:54
心肺血管病杂志(2019年1期)2019-04-22 01:11:58
当代化工研究(2016年9期)2016-03-20 16:22:19
中国塑料(2015年8期)2015-10-14 01:10:49
无机化学学报(2014年12期)2014-02-28 17:34:01
西南军医(2014年4期)2014-01-19 13:54:28
