
含5-(2-吡嗪基)-1H-四氮唑配体的新型锶配合物的合成及固态荧光性质
2024-04-27 09:48赵成兄杨周吉
合成化学 2024年4期
赵成兄, 杨周吉, 孙 赞*
(1. 青海民族大学 化学化工学院,青海 西宁 810007; 2. 青藏高原资源化学与生态环境保护国家民委重点实验室(青海民族大学),青海 西宁 810007)
金属配合物因其组成的多样性、结构的可调控性、良好的热稳定性及不饱和的金属位点等优点,使其在发光[1-6]、催化[7-9]、传感[10-11]和电化学[12]等方面有着广泛的应用前景。吡嗪基四氮唑类配体分子内不仅含有大共轭体系,可以充当好的σ给电子体和π受电子体,还含有能提供多个配位原子的吡嗪环和四氮唑环,可以用来构建具有新颖结构的配位聚合物。影响配位聚合物结构及性质的因素有很多,包括金属离子的本性、配体的配位能力及配位方式、反应体系的pH和反应物的配比等[13-14]。与过渡金属配位聚合物相比,碱土金属离子具有更高的配位数和更灵活的配位几何构型,这使其容易形成多种多样的结构,有些碱土金属配合物还表现出特殊的性质[15]。
因此,本文选择以2-吡嗪基四氮唑(HL)、胡椒酸(HL1)(图1)作为配体,与SrCl2通过溶剂热法合成新型的Sr配合物{[Sr(L)2]·2H2O}n(I),并通过EA、 SXRD、 PXRD、 IR和TGA进行结构表征,研究了配合物I的固态发光行为。
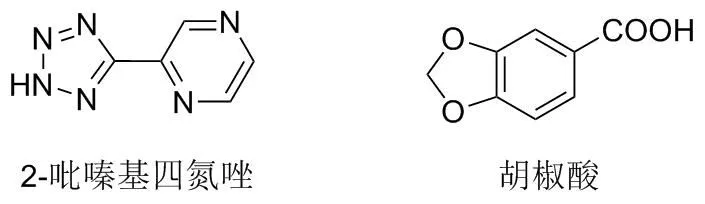
图1 配体的结构
1 实验部分
1.1 仪器与试剂
FTS-3000 FT-IR型傅利叶红外光谱仪; EL-III型元素分析仪; Rigaku Ultima IV型X-射线粉末衍射仪; Rigaku Saturn型X-射线单晶衍射仪; DHG-9240A型电热恒温鼓风干燥箱; Sartourius SQP型电子天平; NETZSCH TG 209型热重分析仪; FLS1000型荧光分光光度仪。
六水合氯化锶(SrCl2·6H2O)(天津希恩思生化科技有限公司),N,N-二甲基甲酰胺(DMF)(天津市凯通化学试剂有限公司),2-吡嗪基四氮唑(河南阿尔法化工有限公司),胡椒酸(武汉丰竹林化学科技有限公司)。所用试剂皆为分析纯。
1.2 配合物{[Sr(L)2]·2H2O}n(I)的合成
将26.6 mg(0.1 mmol)六水合氯化锶、14.8 mg(0.1 mmol, HL) 5-(2-吡嗪基)-1H-四唑、16.6 mg(0.1 mmol, HL1)胡椒酸、3 mL DMF和5 mL H2O同时加入到25 mL烧杯内,超声10 min,待分散均匀后,转移到容积为25 mL的带聚四氟乙烯内衬的不锈钢水热反应釜内,在120 ℃恒温反应24 h后,自然冷却至室温,得到无色块状晶体,产率31%;元素分析,C10H10N12O2Sr,计算值(实测值)%: C 28.74(28.19); H 2.41(2.12); N 40.22(40.63); IRv:3539(m), 3074(w), 1844(w), 1629(m), 1583(m), 1508(m), 1481(w), 1449(m), 1401(w), 1769(m), 1725(m), 1645(m), 1592(s), 1512(m), 1438(m), 1381(s), 1336(m), 1291(m), 1223(s), 1155(m), 1118(m), 1040(s), 869(m), 815(s), 761(s), 644(s), 591(w),556(m), 479(w), 412(w) cm-1。
1.3 X-射线衍射实验
选取尺寸大小合适的无色晶体置于CrysAlisPro单晶衍射仪上,用经石墨单色器单色化的Mo-Kα(λ=0.071073 nm)射线作为光源,在293 K下收集衍射点。配合物以ω-φ扫描的方式收集衍射数据。所有衍射数据使用SADABS程序进行半经验吸收校正。晶胞参数用最小二乘法确定。数据还原和结构解析工作分别使用SAINT和SHELXS-2018程序完成[18]。晶体结构用直接法解出,先用差值函数法和最小二乘法确定全部非氢原子坐标,并用理论加氢法得到氢原子位置,然后用最小二乘法对晶体结构进行精修。主要晶体学数据详见表1,部分键长及键角数据见表2。

表1 配合物I的晶体学以及结构精修参数*
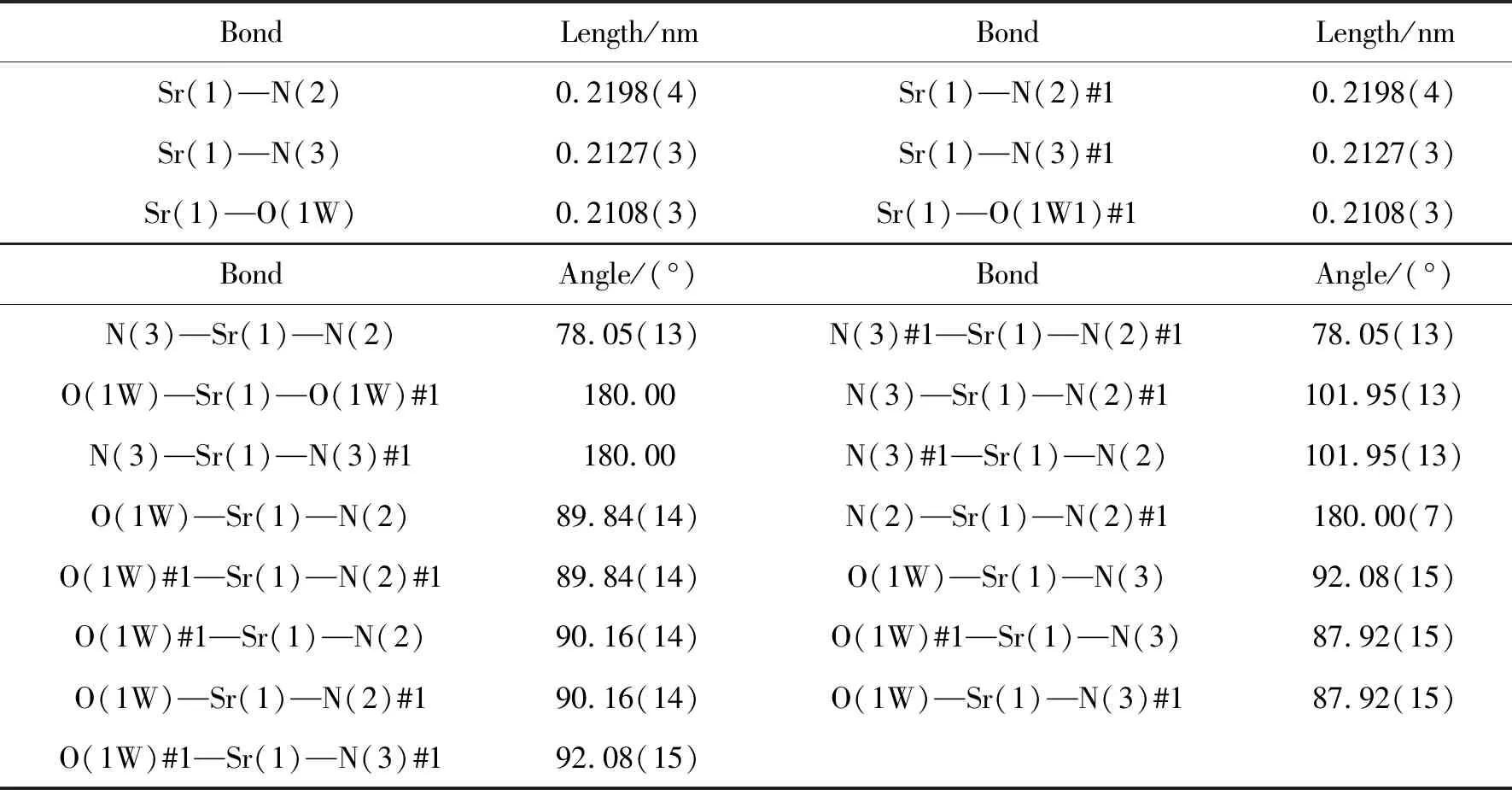
表2 配合物I的部分键长和键角
2 结果与讨论
2.1 配合物{[Sr(L)2]·2H2O}n(I)的晶体结构
单晶衍射数据表明,配合物I属于单斜晶系P21/n空间群,晶胞参数为a=0.61061(4) nm,b=1.14874(7) nm,c=1.07136(7) nm,α=90°,β=105.577(7)°,γ=90°配合物I的结构式为{[Sr(L)2]·2H2O}n。每个不对称单元包含1个独立的Sr、 2个5-(2-吡嗪基)-1H-四氮唑配体和2个配位水分子。在配合物I中,中心金属Sr(II)离子为八面体的配位环境,如图2(a)所示,由来自2个配体分子上的4个N和2个水分子上的2个O组成,而胡椒酸没有参与配位。通过shape软件计算(表3),发现Sr(II)离子的几何构型为略微扭曲的八面体构型,如图2(b)所示,配体的配位模式为螯合配位模式,如图2(c)所示。2个四氮唑配体以反向平行的方式连接SrII形成单核结构。单核之间的氢键相互作用(表4)连接单核形成二维超分子网络结构,如图2(d)所示。

表3 SHAPE软件分析配合物I中Sr(II)离子的几何构型

图2 配合物I中Sr(II)离子的配位环境以及单核结构单元(a), Sr(II)离子的八面体几何构型(b),配体的配位模式(c)和由O—H···N氢键形成的二维超分子结构(d)(对称码: #1 -x, -y+1, -z)
2.2 配合物{[Sr(L)2]·2H2O}n(I)的红外光谱分析
图3为配合物I的红外光谱,可以看出,ν=3365 cm-1为—OH的吸收峰,ν=3076 cm-1为—CH的吸收峰,ν=1446 cm-1、 1414 cm-1、 1357 cm-1、 1274 cm-1、 1224 cm-1、 1150 cm-1和1159 cm-1说明存在—C=C—、 —C=N—的伸缩振动,ν=863 cm-1、 681 cm-1、 756 cm-1和418 cm-1说明有吡嗪环、四氮唑环存在。

ν/cm-1
2.3 配合物{[Sr(L)2]·2H2O}n(I)的X-射线粉末衍射分析
为了验证配合物I是否为单一的纯相,用X-射线粉末衍射(PXRD)对合成的样品进行了测试,所得图谱如图4所示。从图4可以看出,实验测得的XRD谱线与通过单晶数据模拟得到的XRD谱线高度吻合,衍射峰位置一一对应,说明样品中几乎没有杂质,表现出很好的纯度。由此推测实验产品为纯相,验证的样品与所测的结构一致。
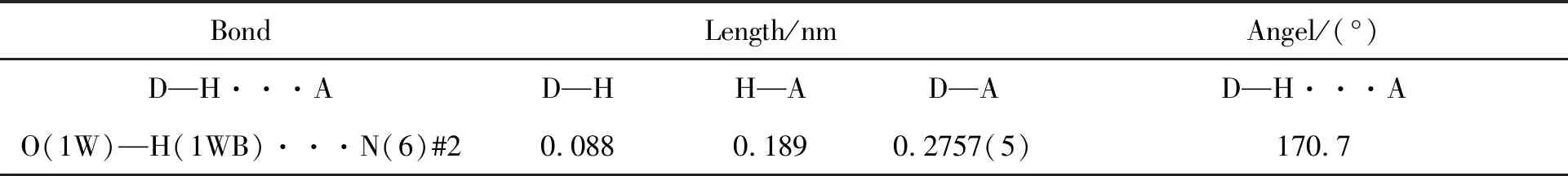
表 4 配合物I的氢键键长和键角*

2θ/(°)
2.4 配合物{[Sr(L)2]·2H2O}n(I)的荧光光谱
图5为配合物I的荧光光谱,可以看出,配合物I具有良好的荧光性能,最大发射波长为450 nm(λex=223 nm),表明配合物I具有良好的荧光性能,是一种潜在的蓝光材料。由于SrII离子的电子构型与稀有气体Kr相同,其电子跃迁所需的能量较高,所以SrII离子与配体形成的复杂分子在紫外光激发下,电子跃迁基本上局限于L→L*跃迁。因此,配合物经紫外光激发后发射的荧光基本上是由配体L*→L的电子迁移引起的,配合物I的荧光光谱是仅受中心离子微扰的配体的荧光光谱[19]。
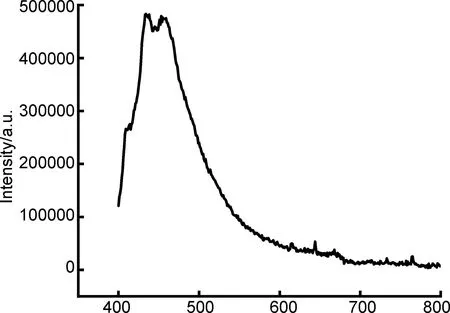
λ/nm
2.5 配合物{[Sr(L)2]·2H2O}n(I)的热重分析
在氮气气氛中,升温速率为10 ℃/min, 25~800 ℃内对配合物I进行热稳定性能测试(图6)。从图6可发现配合物I在25~125 ℃之间保持结构的稳定; 125~225 ℃为第1次失重8.89%(理论值为8.61%),可归因于损失了2个配位水分子;在325~540 ℃的温度范围内,发生第2次失重,这是由于随着温度的升高,配合物的结构开始分解;直到540 ℃左右趋于稳定,最终残余物可能是SrO,残留量为23.48%(理论值为24.79%)。

Temperature/℃
本文采用溶剂热法在水和DMF的混合溶剂中合成了锶配合物I{[Sr(L)2]·2H2O}n,结构分析表明:配合物I属于单斜晶系,晶胞参数为a=0.61061(4) nm,b=1.14874(7) nm,c=1.07136(7) nm,α=90°,β=105.577(7)°,γ=90°。 Sr中心采取六配位的八面体几何构型,通过分子间氢键形成二维超分子结构。荧光光谱数据表明:锶配合物具有良好的荧光性能,是一种潜在的蓝光材料。
猜你喜欢
陶瓷学报(2021年3期)2021-07-22
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
新世纪智能(数学备考)(2019年9期)2019-10-16
当代陕西(2019年6期)2019-04-17
物理学进展(2017年1期)2017-02-23
大学化学(2015年5期)2015-09-18
中国酿造(2015年4期)2015-01-26
火炸药学报(2014年1期)2014-03-20
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年4期)2014-02-28
