
一种基于real-time PCR 技术的TTV 检测方法的建立及应用
2024-04-24 12:41贾毅博王高玉邓宛心林彩云陈运春尹飞飞
海南医学院学报 2024年7期
贾毅博,王高玉,邓宛心,林彩云,杨 华,陈运春,尹飞飞
(1.海南医学院公共卫生与全健康国际学院,海南 海口 571199;2.海南医学院-香港大学热带传染病联合实验室,海南 海口571199;3.海南省妇女儿童医学中心,海南 海口 570206;4.海口市中医医院,海南 海口 570206)
TTV 病毒(Torque Teno virus, TTV)属于指环病毒科(Anelloviridae), α 指环病毒属(Alphatorquetenovirus),是一种无囊膜,单链环状的DNA病毒。该病毒基因组约为3.8 kb,包括四个开放阅读框(open reading frame,ORF)以及高变区(hypervariable region,HVR)和N 末端精氨酸片段[1,2]。TTV 病毒最早于1997 年在日本一名病因不明的急性输血后肝炎患者体内被首次发现并报道[3]。TTV 是血液病毒组中最常见的病毒种类,其致病性目前仍然不明确。部分研究表明TTV 和肝炎、呼吸道疾病、肿瘤以及自身免疫性疾病等相关,但是近年来的研究认为其很少会直接导致人和动物感染性疾病的发生[4,5]。TTV 的致病性及复制机理等研究面临最主要的问题是尚未有灵敏的体外细胞培养系统和动物感染模型。该病毒宿主范围十分广泛,除了人类,目前已经在多种动物中被发现,包括猪、骆驼、马、猫、狗、獾、海狮和非人灵长类动物等[3]。流行病学研究结果表明,TTV 在全球范围的人群中广泛存在,在俄罗斯、日本和巴基斯坦等健康人群中流行率高达90%以上[6-8]。
近年来TTV 作为评估人体免疫功能的重要指标受到广泛关注,尤其在器官移植患者中已进入临床应用[9-11]。尽管TTV 病毒具有广泛的临床重要性,但目前还没有广谱、便捷、高效和灵敏度高的TTV 检测方法。当前针对TTV 的检测方法主要是基于分子生物学的诊断方法,主要有聚合酶链式反应(Polymerase Chain Reaction,PCR)、实时荧光定量PCR(Real-time PCR)等技术。由于TTV 基因组的高度多样性和变异性,不同基因型之间的变异程度最高可达到50%以上,常见的PCR 检测方法只能针对部分TTV 亚型进行检测,且检测方法灵敏度不够,容易出现漏检等问题[12,13]。在2001 年,Pistello 等[14]针对特定地区流行的TTV 亚型,基于TTV 基因组的ORF1 区域,开发了一种新的realtime PCR 检测方法。这种方法虽然能够特异性地识别某些亚型,但由于选择的扩增区域的局限性,对其他TTV 亚型的检测能力有所不足。与此同时,Maggi 等[15]研究者根据TTV 基因组的UTR(untranslated regions,UTR)区域,设计了新的引物和探针,进而建立了另一种real-time PCR 检测方法,尽管这一方法在检测范围上取得了一定提升,但其检测灵敏度仍然较低,仅为1×103copies/μL。在2017 年,法国的研究人员推出建立了一种用于检测TTV 的标准化real-time PCR 方法,且在健康志愿者、捐赠者和肾移植接受者中进行了测试,但是其只可以对12 种TTV 亚型进行检测[16]。而酶免疫测定(enzyme immunoassay,EIA)和酶联免疫吸附实验(enzyme-linked immuno-sorbent assay,ELISA),尽管可以用于检测TTV 抗体,但由于IgM 和IgG 抗体之间时间重叠较大,很难区分当前感染和既往感染[13]。这些问题限制了对TTV 感染的深入研究和其作为疾病标志物的应用潜力。鉴于现有的TTV 检测方法存在的诸多局限性,本研究建立了一种具有更高的灵敏性和特异性的real-time PCR 方法,为揭示TTV 在多种疾病过程中的作用提供技术支持。
1 材料与方法
1.1 实验试剂及引物设计合成
从海南省妇女儿童中心收集30 份临床血液样本,经过3 500 r/min,3 min 离心后,分装保存于- 80 ℃冰箱备用。 血清样本病毒DNA 使用QIAamp DNA Mini Kit(德国,QIAGEN 公司)进行提取;PCR 扩增使用KOD One™ PCR Master Mix,(日本,TOYOBO 公司产品);Real-Time PCR 反应使用Premix Ex Taq™(Probe qPCR),(宝生物工程(大连)有限公司);柱离心型DNA 凝胶回收与纯化试剂盒和高纯质粒小量提取试剂盒,均为天根生化科技(北京)有限公司产品。
使用BioEdit 软件对ICTV 所公布的22 种不同亚型的TTV 序列进行比对(参考序列基因号:AB008394、AB049608、AY666122、AB041957、AF345523、 AF435014、 AF261761、 DQ187006、AB064607、 AF345526、 AB037926、 AB028668、AX025830、AX025718、AB025946、AB060594、AF348409、 AB060597、 AB041959、 AB041958、AB038621、KJ082064),在此基础上,设计一套可针对已知所有TTV 亚型的检测引物以及探针。选择文献报道目前应用较为广泛的5 套引物(表1)用于本研究质粒的扩增以及对real-Time PCR 方法检测能力的对比研究。以上所有引物及探针均委托由生工生物工程(上海)股份有限公司合成。
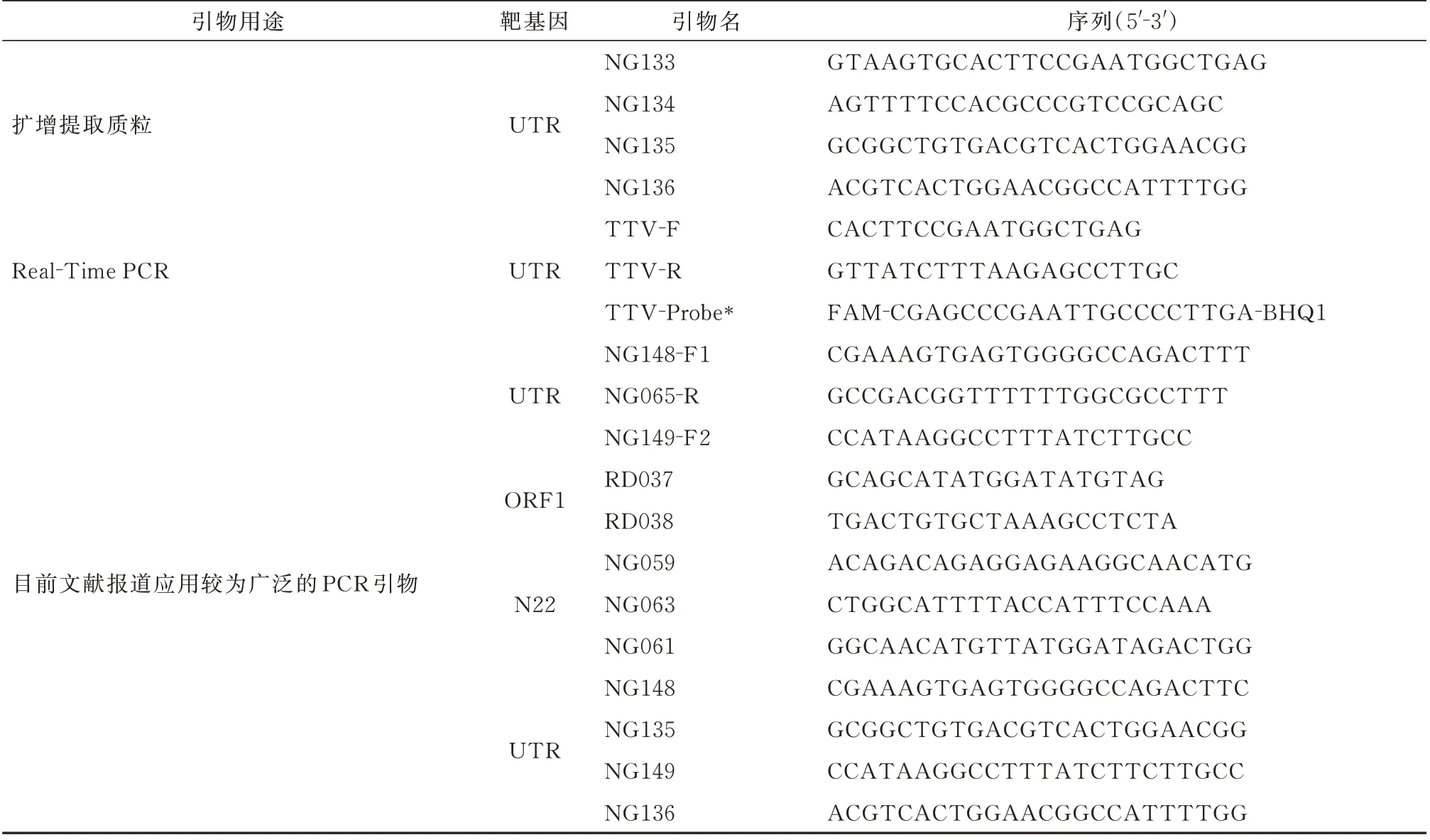
表1 标准品制备及检测相关引物和探针序列Tab 1 Sequences of primers and probes used in the preparation of standard samples and detection
1.2 标准品的制备
以临床血液样本提取的TTV 病毒DNA 为模板,分别利用两对引物NG133/NG136、NG134/NG135 进行巢式PCR。反应体系为:KOD One™PCR Master Mix,12.5 μL,上、下游引物,引物浓度为10 μmol/L,各1 μL;DNA 模板,1 μL;灭菌双蒸水,9.5 μL。PCR 反应条件为:98 ℃,2 min;98 ℃,10 s;60 ℃,10 s;68 ℃,30 s;35 个循环;68 ℃,5 min;4 ℃终止反应。扩增产物为3 354 bp,经琼脂糖凝胶电泳后,切取目的片段并使用试剂盒回收纯化。随后与pLB vector 连接后转化到DH5-α 感受态细胞中。在含氨苄青霉素的培养板上37 ℃培养16 h,挑取单个菌落,增菌后用质粒小量提取试剂盒提取质粒进行PCR 鉴定,测序验证。
以提取的含有TTV 片段的质粒为标准品,用NAS-99 微量分光光度计测定其浓度,根据阿弗加德罗数将浓度换算为拷贝数,然后用灭菌双蒸水依次进行梯度稀释至1×107copies/μL、1×106copies/μL、1×105copies/μL、1×104copies/μL、1×103copies/μL、1×102copies/μL、1×101copies/μL,分装后置于-20 ℃保存备用。
1.3 real-time PCR 反应条件的优化
为了得到更高的荧光增幅并使反应有最小的循环数(Cycle threshold,CT),对real-time PCR 的退火温度进行梯度优化,即从56 ℃开始,每次提高2 ℃的退火温度,直至60 ℃截止;同时为提高realtime PCR 反应中TTV 的扩增效率,我们对反应体系中的引物和探针终浓度由0.1 μmol/L 开始,每次增加一倍,直至终浓度为0.4 μmol/L 截止。
1.4 real-time PCR 敏感性、稳定性试验及标准曲线的建立
将倍比稀释浓度在1.0×101~1.0×107copies/μL 之间的质粒标准品,按优化后的反应体系以及条件进行PCR 扩增检测,最终反应体系为25 μL,主要为Premix Ex Taq(Probe qPCR)(2×)12.5 μL,上游引物TTV-F、下游引物TTV-R 以及特异性探针TTV-P 各0.5 μL,各浓度梯度模板2 μL、灭菌双蒸水9 μL;并设置以灭菌双蒸水为模板的阴性对照,观察该方法的敏感性;扩增条件均为优化后最佳反应,即95℃,30 s;95℃,5 s;60℃,30 s;40 个循环。标准曲线由BioRad CFX96 rM Real-Time PCR Detection System 扩增仪自动生成,X 轴代表每个浓度标准品模板中所含病毒起始含量以10 为底的对数,Y轴代表各个梯度质粒标准品扩增时达到阈值所需的循环数,反复实验3 次评估试验稳定性以及引物最低反应拷贝数。
1.5 与目前文献报道应用较为广泛的PCR 检测方法的对比
使用文献报道的4 套PCR 扩增引物组合(见表1)和本研究设计的real-time PCR 方法,分别对收集的30 份临床血清样本进行检测。本研究所使用的引物相对位置(图1),各引物组合PCR 反应条件以及扩增长度(表2)。
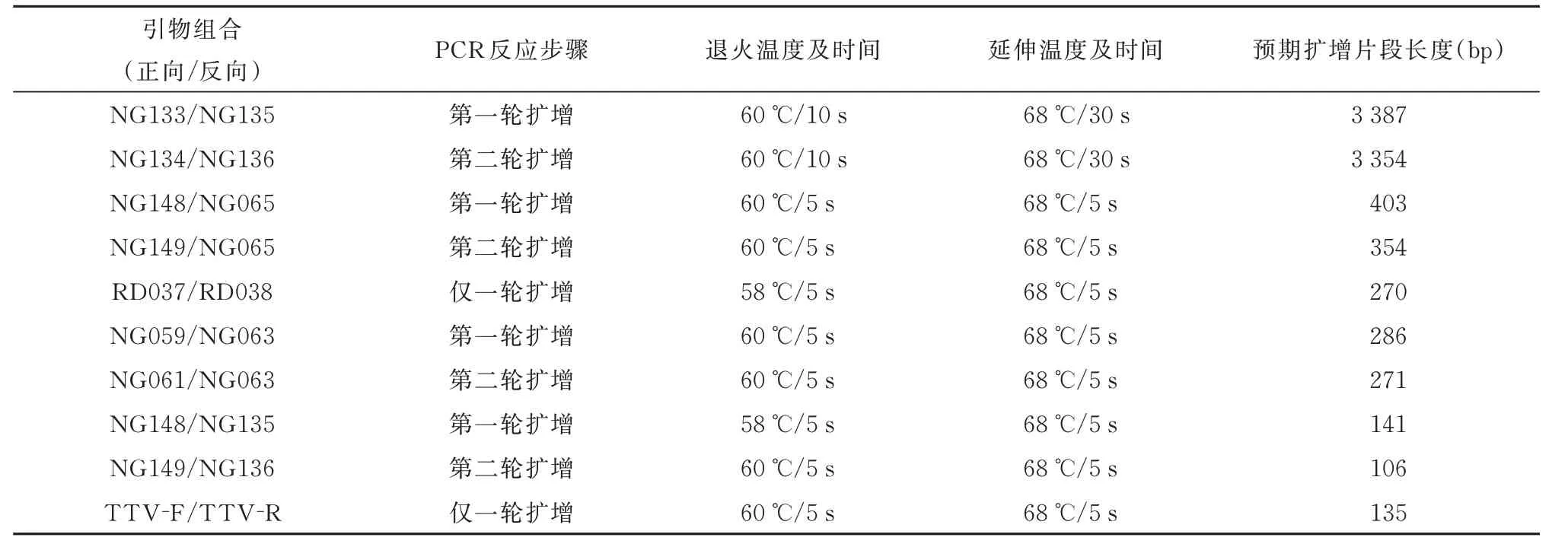
表2 各引物组合PCR 反应条件、扩增长度Tab 2 PCR conditions and amplification length of each primer combination
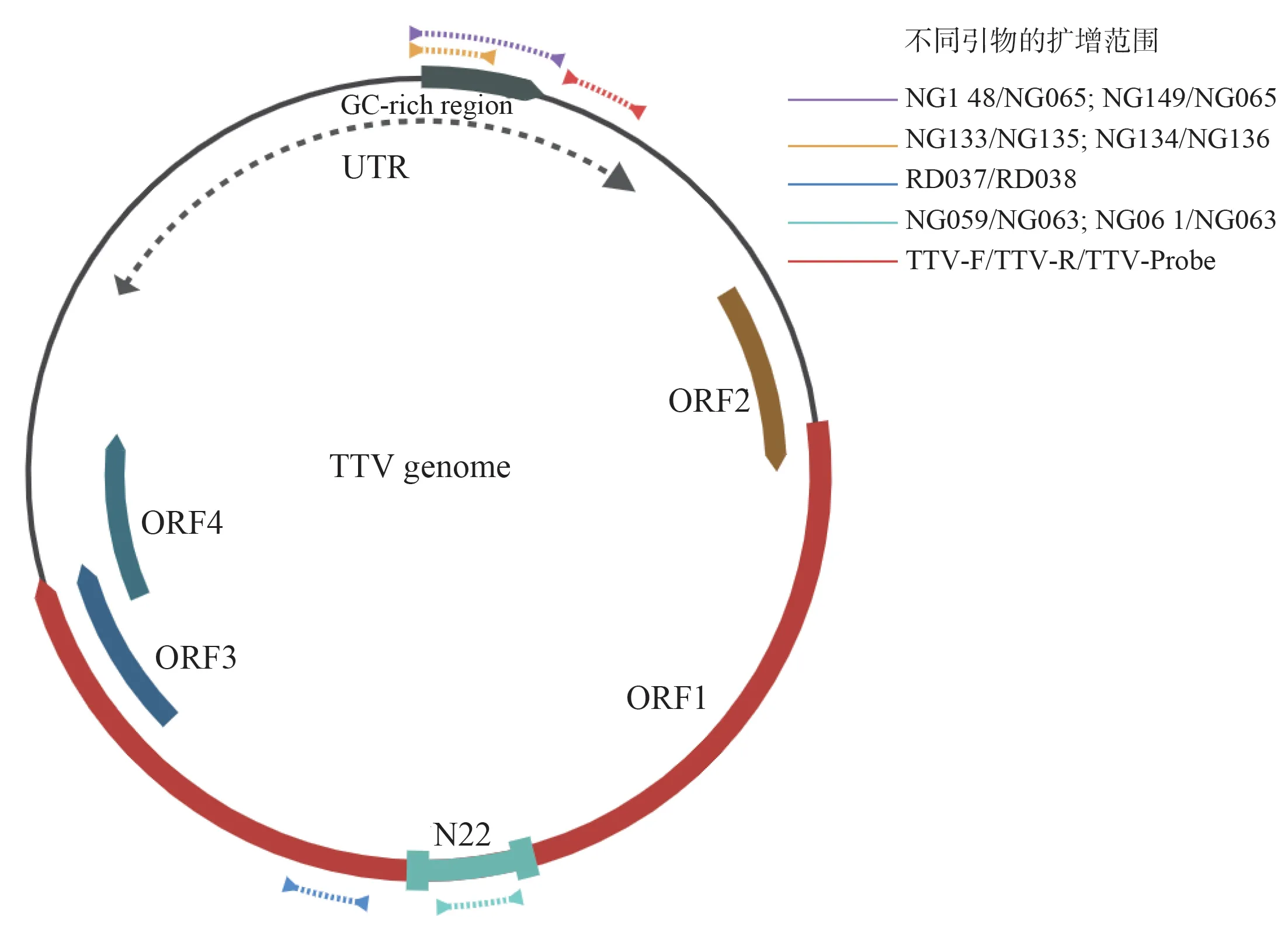
图1 TTV 基因组结构以及本研究所使用的引物相对位置示意图Fig 1 The genome structure of TTV and the primers' relative positions used in this study
2 结果
2.1 TTV 病毒Real-time PCR 检测方法的建立
质粒标准品的制备:以TTV 阳性血清样本提取的病毒DNA 为模板,经巢氏PCR 扩增,得到大小为3 354 bp 的目的片段(见图2)。将此特异性片段与pLB 载体连接,并进行测序,测序结果经BLAST分析,结果显示与TTV 病毒DNA 依赖性RNA 聚合酶序列(GenBank accession no.AB026345)的一致性为98.25%,表明该质粒中插入基因为TTV 病毒基因,因此用于标准曲线的构建。

图2 TTV 阳性血清样本扩增结果电泳图Fig 2 Electrophoresis image of amplification results from positive serum samples of TTV
2.2 Real-Time PCR 反应体系及条件的优化
为了得到更高的荧光增幅并使反应有最小的循环数(CT),对real-time PCR 的各项条件进行了摸索,其中,对反应体系的优化实验则主要为反应体系中引物、探针使用的终浓度进行优化筛选,取制备的终浓度为107copies/μL~101copies/μL 的系列标准物质,分别作为DNA 模板,实验中的引物和探针的配制浓度均为10 μmol/L,添加量分别使用1 μL、0.5 μL 和0.25 μL,放入Real-Time PCR 仪中使用相同的PCR 扩增程序(比如95 ℃预变性30 s;95 ℃,5 s;58 ℃ ,30 s;40 个循环),进行Real-time PCR 扩增,得到各自不同的扩增效率(E)、相关系数(R2)以及斜率,具体结果如(表3)所述,综合比较可见引物和探针终浓度不同,扩增效率差异显著,以添加量后的终浓度为0.2 μmol/L 最优。最终优化的25 μL 的PCR 反应体系:PCR 管中依次加入2×PCR 预混液12.5 μL,10 μmol/L 引物和探针各0.50 μL,DNA 模板2 μL,去离子水9 μL,充分混匀。
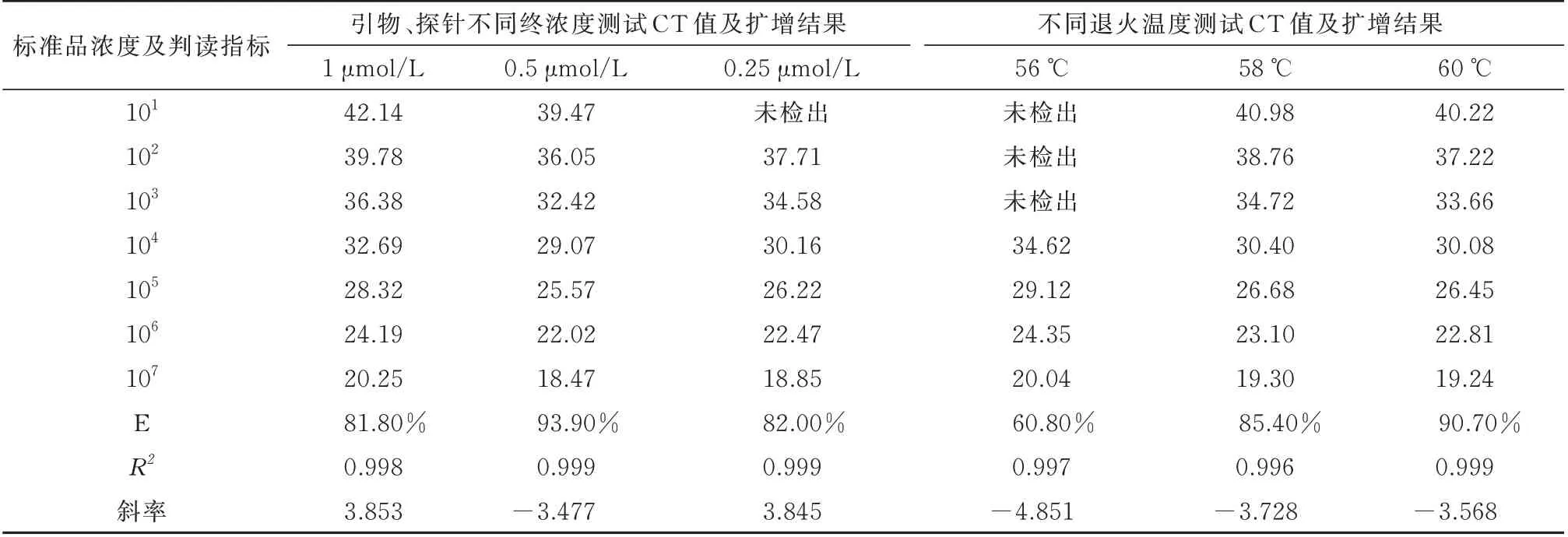
表3 Real-Time PCR 反应体系及条件的优化Tab 3 Real-time PCR system and optimization of conditions
为进一步提高real-time PCR 的扩增效率,我们对PCR 扩增程序的优化实验包括对各个反应参数的优化筛选,如变性温度、变性时间、退火温度、退火时间、延伸温度、延伸时间和循环数。经验证:本研究设计的real-time PCR 扩增程序可以优选二温度点法,即除变性温度外、退火与延伸温度可合二为一,该扩增效率与常规的三温度点法(变性温度、退火温度、延伸温度)相差不大。其中,主要对退火/延伸的温度进行摸索,分别选择56 ℃、58 ℃和60 ℃这3 个温度进行退火/延伸温度的最后优化验证,根据上述已经优选的PCR 扩增程序,取制备的终浓度为107copies/μL~101copies/μL 的系列标准品,分别作为DNA 模板、添加量为2 μL,引物和探针(浓度均为10 μmol/L)的添加量为优选结果0.5 μL,反应体系其他组份参考上述对反应体系的优化筛选结果,放入real-Time PCR 仪中,PCR 扩增程序的参数条件除退火/延伸温度参数以外,其他参数固定,进行real-Time PCR 扩增,得到扩增效率(E)、相关系数(R2)以及斜率,具体结果如下(表3)所述,综合比较可见退火/延伸温度不同,扩增效率差异显著,以60 ℃的反应结果最优。最终确定优化后的PCR 扩增程序为:95 ℃预变性30 s;95 ℃变性5 s,60 ℃退火延伸30 s,40 个循环。
2.3 Real-Time PCR 的灵敏度检测
根据上述建立并优化验证的TTV real-time PCR 检测方法,对终浓度为107copies/μL~101copies/μL 的标准品进行检测,以每个浓度标准品的PCR 扩增的相对荧光单位为纵坐标,以PCR 扩增程序中的循环数为横坐标,得到扩增曲线;再以每个浓度标准物质终浓度的常用对数(lgC)为横坐标,以循环次数为纵坐标,获得TTV 多亚型PCR 检测的标准曲线如下(图3)所示。在多次实验中,当标准品的终浓度为101copies/μL 时均可见PCR 扩增、且扩增效率E 均大于90%,同时,我们对该定量产物通过凝胶电泳系统进行检测,结果显示初始标准品浓度在107copies/μL~101copies/μL 时,扩增产物均可见到明显的条带(图4),但100copies/μL 无法成功检测,因此确定本研究建立的TTV 多亚型检测方法最低检测限101copies/μL。
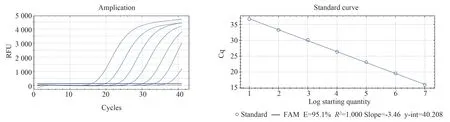
图3 TTV 多基因亚型real-time PCR 检测方法的标准曲线Fig 3 Standard curve of a real-time method for detecting the polygenic subtypes of TTV
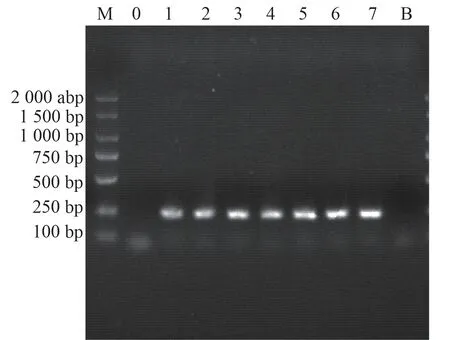
图4 对不同浓度的标准品扩增产物凝胶电泳图Fig 4 Gel electrophoresis image of amplification products from standards at different concentrations
2.4 Real-Time PCR 的稳定性检测
采用本实验中开发的Real-Time PCR 方法对105copies/μL 的标准品进行了6 次重复的批内稳定性测试(图5),重复实验的CT 值分别为26.10、26.22、26.27、26.34、26.34 以及26.67。平均CT 为26.32,标准差为0.19,变异系数为7.22%。结果显示出良好的重复性。
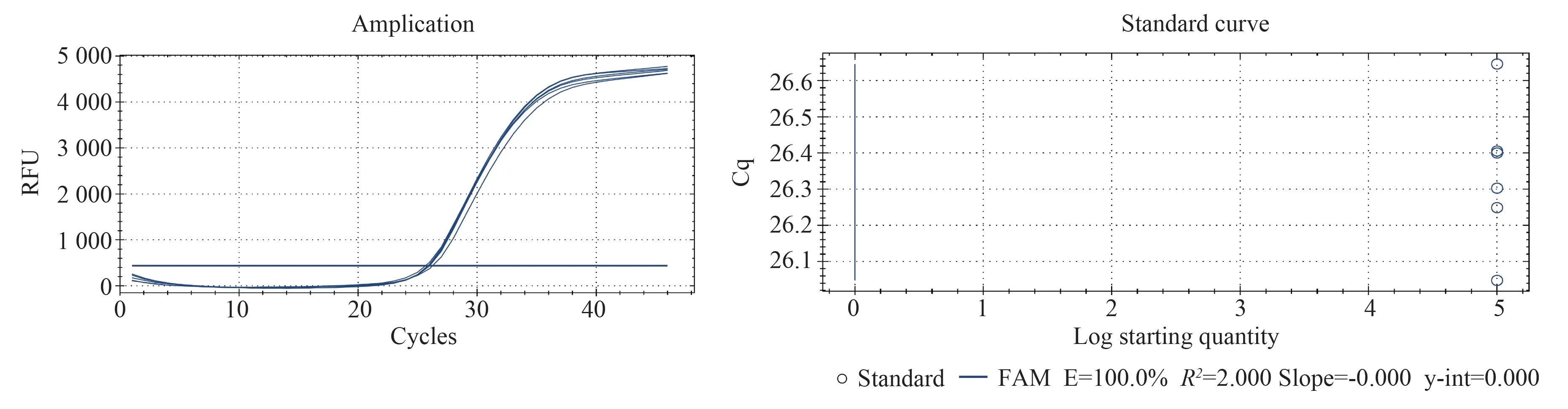
图5 TTV 多基因亚型real-time PCR 检测方法组间差异实验分析Fig 5 Experimental analysis of within-group variations in the real-time PCR method for multiple genotypic subtypes of TTV
2.5 与目前文献报道的PCR 检测方法对比结果及特异性验证
本文选择Okamoto 等[17]报道的且目前被多个研究所使用的4 套引物与本研究建立的方法,同时对30 份临床血液样本以及稀释为103~101copies/μL 的TTV 质粒进行检测,为减少实验误差,将每个浓度的质粒,在同一批次内进行了4 次扩增。随后对所有扩增产物进行Sanger 测序,测序结果在NCBI(National Center for Biotechnology Information)数据库进行Blast 比对。检测结果如下(表4)所示,统计分析发现具有显著差异(χ2=51.05,P<0.01)。无论是对临床样本还是不同病毒载量的质粒进行检测,本研究所建立的方法灵敏度远远高于文献中报道的其他方法;同时测序结果分析表明,本方法所扩增出的30 份阳性样本产物均为TTV,特异性为100%。
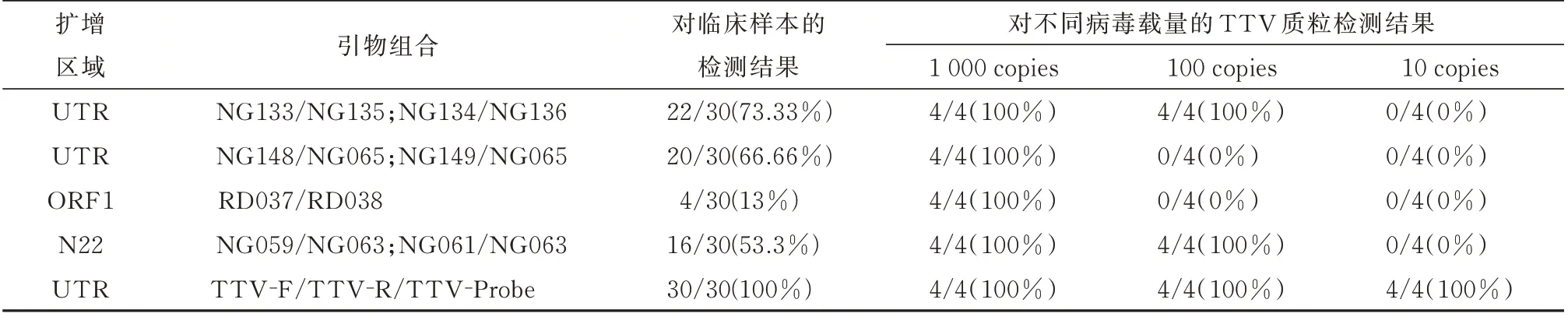
表4 用不同区域设计的引物对临床样本及不同病毒载量的TTV 质粒检测结果Tab 4 Detection results of clinical samples and TTV plasmids with different doses of virus using primers designed in different regions
3 讨论
自1997 年TTV 被发现以来,由于其人群感染率高和显著的遗传多态性的特点,受到了研究者的广泛关注[1,18]。然而由于缺乏灵敏的体外细胞培养系统和动物感染模型,目前对TTV 致病性及其与特定疾病间的相互关系尚未明确。研究表明,在免疫抑制状态下,TTV 的复制活动明显增强。在健康人体内,每天可以产生超过3.8×10¹⁰ copies/mL 的病毒粒子,其中超过90%的病毒粒子被免疫系统清除。与健康个体相比,免疫功能较弱的患者体内的TTV DNA 载量更高[16]。因此,TTV 血液中的水平可能反映了患者的免疫状态,高TTV DNA 载量表明过度免疫抑制和感染风险,而低载量则可能指示免疫抑制不足和排斥反应的风险[10]。近年来,研究人员已经开始探索TTV 在人类血浆中的病毒载量,试图将其与免疫状态、感染或器官移植后的排斥反应相关联。TTV 正日益被认为是评估整体免疫能力、预测移植后免疫相关不良事件、并最终制定免疫抑制治疗的一个重要生物标志物[9,19-21]。最近的研究已经明确了使用TTV 监测多种免疫抑制疾病患者的可行性,包括HIV 患者、实体器官移植患者、造血干细胞移植后的患者以及类风湿性关节炎治疗患者[22-24]。此外,TTV 还可以预测严重COVID-19 患者的院内感染风险,以及指示慢性阻塞性肺病患者的肺功能及疾病的严重程度,显示了其在临床应用中的重要价值[25,26]。
2020 年国际病毒分类委员会(ICTV)更新的数据将TTV 分为22 个基因型,不同的TTV 分离株之间的核苷酸差异超过30%[27]。此外,TTV 被分为5 个亚群,各亚群之间的差异超过50%。在基于靶向扩增的real-time PCR 检测方法建立过程中,病毒DNA 片段的选择对PCR 检测灵敏度有显著影响[28-30]。自TTV 发现以来,ORF1 的N22 区域在巢氏或半巢式聚合酶链式反应中被广泛应用[31]。但是由于此区域序列的复杂多变,因此该区域所设计的引物很难扩增所有的TTV 分离株。近期对TTV 的进一步研究发现,TTV 基因组的UTR 区域由于其更高的保守性,更加适合设计检测引物,在UTR 区域设计的引物已被证明可检测出目前公认的大多数TTV 亚型[30,32]。与ORF1 区域设计的引物相比,UTR 区设计的引物,极大的提高了TTV DNA 在不同样本中的检出率。在Takahashi等[33]的研究中,将UTR 区域所设计的引物与ORF1区域所设计的引物进行对比发现,同样的样本,前者的TTV 检出率为92%,而后者的TTV 检出率只有23%。Irving 等[34]的研究中也报道了这一情况,将UTR 区域所设计的引物与ORF1 区域所设计的引物进行对比发现,前者的检出率为50%,而后者只有9%。同时,在Itoh 等[29]的研究中,将UTR 区域所设计的引物与ORF1 区域所设计的引物进行对比发现,前者的检出率为95%,而后者只有20%。与此同时,有研究认为,虽然UTR 区域较ORF1 区域所设计的引物,其对TTV 的检出率会极大提高;但可能会由于研究者设计位点选择不够精确的原因,而出现所设计的引物更加偏向于某一个型别的TTV 且对其他型别不够灵敏的现象[30]。目前以TTV 为标记物来评估器官移植等患者的免疫功能,不只需要对TTV 进行检测,还需要对其进行精确定量[35]。然而,由于应用于TTV 定量的不同的real-time PCR 方法检测结果间存在差异,因此,对TTV 病毒载量的检测需要进行严格的评估和比较。一般来说,使用基于UTR 区域的检测方法,其灵敏度较基于ORF 区域的检测能力高10~100 倍[17,33]。现阶段所报道的Real-time PCR 测定的检测下限约为102copies/μL~103copies/μL,且该检测值仅可以用于测定评估的特定TTV 基因型[36,37]。面对这种挑战,本研究开发的Real-time PCR 方法无疑提供了一个可行的工具。笔者分析了具有序列高变性的多个亚型的TTV 引物设计的靶基因序列,筛选到本研究所述能广谱检测到多个亚型TTV 的引物和探针组合物。此方法相对于已公开的其他引物组合物,检测覆盖范围更广,检测灵敏度高达101copies/μL。通过核酸测序分析验证,本研究检测TTV 的阳性符合率为100%,对TTV 检测的特异性更高。
本研究的主要局限在于为了能够检测更多的TTV 亚型,real-time PCR 检测方法的引物选取了高度保守的UTR 区域的保守片段。因此,虽然本realtime PCR 检测方法能够检测到目前已知大部分基因型的TTV,但不能用于区分TTV 的亚型。未来的研究应考虑开发既能覆盖广泛基因型的检测方法,又能对TTV 的不同亚型进行更区分的检测方法。
综上所述,本研究采用基于TaqMan 探针的real-time PCR 检测方法,检测灵敏性高、覆盖基因型范围广,检出率高,尤其对于TTV 病毒载量较低的情况下能够实现准确的定量检测,为探索TTV 作为疾病生物标志物的潜在应用提供重要的更全面的技术支持。
作者贡献度说明:
贾毅博:实验操作并撰写论文;王高玉、邓宛心:负责样本收集与数据整理;林彩云、杨华、陈运春:负责论文审阅与修改;尹飞飞:全程参与、指导实验操作并修改论文。
所有作者声明不存在利益冲突关系。
猜你喜欢
食品科学(2018年10期)2018-05-23
现代检验医学杂志(2016年3期)2016-11-15
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国病理生理杂志(2015年8期)2015-12-21
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年30期)2015-03-01
中国当代医药(2015年9期)2015-03-01
物理实验(2015年9期)2015-02-28
西南军医(2015年6期)2015-01-23
癌变·畸变·突变(2014年2期)2014-03-01
