
分子筛型声学增强材料的储存耐候性能
2024-03-25 02:42孟庆华
高等学校化学学报 2024年3期
孟庆华,史 超
(1. 上海交通大学化学化工学院, 上海 200240; 2. 盐城工学院纺织服装学院, 盐城 224051)
随着多媒体设备的发展,微型扬声器被广泛应用在声学设备中. 然而,由于空间容积限制,微型扬声器的低频率声学性能很难得到提升. 在包括发声装置、 壳体和后腔的扬声器装置中,填充声学增强材料(AEM)是一种改善低频声学性能的有效方法[1~4]. 声学增强材料,如活性碳[5~7]、 介孔硅材料[8]和沸石分子筛[9,10]等可放置在扬声器后腔中,用于提升低频声学性能. 扬声器后腔中的声学增强材料通过对气体的吸附-储存-释放过程,实现后腔显著的虚拟增大效果,改变扬声器装置的声顺性,进而使扬声器达到更优异的声学效果[10].
发声装置的谐振频率(f0)是扬声器重要的声学性能指标,在实际应用中,发声装置的f0过高会造成低音性能降低、 失真等声音性能问题. 通过对发声装置和电子产品的结构进行改进,或者增设其它声学辅助器件,可降低发声装置的f0,进而提高发声装置的声学性能. 发声装置单元的f0可用下式表示:
式中:f0(Hz)为发声装置单元的谐振频率;mms(g)为发声装置单元的质量;Cms(mm/N)为发声装置单元的等效声顺性.
将发声装置单元装配到发声装置的腔体或电子产品腔体中后,其谐振频率f01用下式表示:
式中:Cma(mm/N)为发声装置的腔体容积的空气声顺性.
将声学增强材料放入发声装置的腔体中后,发声装置的谐振频率f02用下式表示:
式中:a为在腔体中加入声学增强材料后的容积被等效扩大的倍数.
在发声装置的腔体中加入声学增强材料后,发声装置的谐振频率偏移值(ΔF0)用下式表示:
在扬声器中常使用多孔物质作为声学增强材料. 当外界声压增大时,声学增强材料可吸附扬声器腔体中的空气,吸附的空气体积通常大于声学增强材料自身的体积,从而实现扬声器腔体的容积被等效扩大的效果. 活性炭是一种具有大比表面积及宽孔径分布的多孔材料,可用于增大扬声器后腔声学体积,但强的吸水性会影响其声学增强效果[5]. 介孔硅是一种以硅和氧为主要构成元素的介孔材料,也可用于增大扬声器后腔体积,充分填充在扬声器后腔可有效降低谐振频率(ΔF0≤112 Hz)[8],但其声学改善效果仍有待提升. 与活性炭和介孔硅结构不同,沸石分子筛是一类具有规则孔道的无机微孔晶体材料[11,12]. 根据拓扑结构的不同,微孔孔道维数从0维~3维(以氩气分子为探针分子,分子动力学直径为0.34 nm),且多维度孔道之间相互贯穿或独立存在. 具有高维孔道的沸石分子筛材料对氮气具有吸附-脱附能力. 将沸石分子筛应用于微型扬声器系统中可起到吸附和储存空气的作用. 当微型扬声器体系中的压力增大时,空气分子被吸附、 储存在分子筛内. 随着压力的降低,空气分子得到释放. 对空气分子的吸附-储存-释放过程可降低微型扬声器系统振动的刚度,最终提升微型扬声器系统的低频声压级性能和声顺性,有效降低谐振频率(ΔF0≥150 Hz)[9].
在扬声器的实际应用场景中,分子筛型声学增强材料受到水分子和有机物小分子等因素的影响.目前,在行业和科研领域内,影响分子筛型声学增强材料的谐振频率的具体因素及作用机理还没有明确统一的认识. 为了探究湿度和有机物小分子等对分子筛型声学增强材料谐振频率的影响,本文研究了由不同硅铝比(SAR)分子筛构成的声学增强材料在不同湿度和不同有机物小分子氛围储存后谐振频率的变化规律. 通过对影响声学增强材料谐振频率的具体因素进行分析讨论,为发掘更优异的声学增强材料提供一定的借鉴与指导.
1 实验部分
1.1 试剂与仪器
LUDOX SiO2溶胶(30%),西格玛奥德里奇(上海)贸易有限公司; NaOH(纯度98%)、 四丙基溴化铵(TPABr,纯度98%)、 Al(NO3)3·9H2O(纯度99%)、 乙醇(体积分数95%)和聚丙烯酸酯IV(化学纯),上海泰坦科技股份有限公司; 甲苯和N,N-二甲基丙烯酰胺(DMPA)均为分析纯,国药集团化学试剂有限公司; 自制高纯水(电导率≤0.06 µS/cm).
XRD-6000 型X 射线衍射仪,日本岛津公司[以CuKα射线为入射光源(λ=0.154056 nm),工作电压为40 kV,工作电流为 30 mA,扫描速率为8°/min,扫描范围为5°~40°]; Clarus SQ 8C型顶空-气相色谱-质谱联用仪,美国珀金埃尔默公司(顶空条件: 样品加热温度120 ℃,进样针温度150 ℃,传输线温度150 ℃,保温时间60 min,进样时间0.05 min,瓶压172.37 kPa; 气相色谱条件: 进样口温度250 ℃,进样方式分流20∶1,载气为氦气,载气流速1 mL/min; 质谱分析条件: GC 传输线温度250 ℃,离子源温度230 ℃); ASAP 2020HD 型气体吸附仪,美国Micromeritics 公司[低温氮气测试需样品在120 ℃真空中预处理3 h,-196 ℃下液氮吸附,采用BET方法计算样品比表面积、 BJH方法计算介孔分布、t-plot 方法计算微孔孔容,采用氮气吸附法测定材料常温(25 ℃)下的单层吸附量]; Nicolet iS5 型傅里叶变换红外光谱仪,赛默飞世尔科技(中国)有限公司(以空气为背景进行室温扫描,扫描范围4000~400 cm-1,扫描次数16次); LRHS-504B-LJS型高低温交变湿热试验箱(环境箱),上海林频仪器股份有限公司.
1.2 分子筛的合成
将Al(NO3)3·9H2O(0.0300,0.0125,0.0075,0.0038 和0 mol; 对应质量分别为11.25,4.69,2.81,1.41 和0 g)分别溶于去离子水(28.33 mol,510.52 g)中,向上述溶液中分别加入NaOH(0.24 mol,9.60 g),搅拌均匀. 搅拌下将上述溶液滴加到300.40 g 30%的LUDOX SiO2溶胶(SiO2含量90.12 g,1.5 mol)中,搅拌3 h后,混合液变为乳白色溶胶,室温下搅拌陈化6 h. 将乳白色溶胶置于高压釜中,于150 ℃晶化72 h后取出,在室温下冷却,减压过滤,用蒸馏水洗涤,得到白色粉末. 将该白色粉末置于烘箱中于120 ℃加热6 h,再置于马弗炉中于600 ℃煅烧6 h 以除去模板剂,最后研磨成粉末,即得到MFI_m分子筛(m为晶化产物中的硅铝摩尔比,取整数).
1.3 谐振频率测试
测试谐振频率时不同硅铝比声学增强材料的用量为140 mg,粒径为300~350 µm,标准腔体的体积为1 mL.
1.4 储存气氛对材料声学性能的影响
考察了以不同硅铝比分子筛为主体材料的声学增强材料分别在常温常湿(25 ℃/50%RH)、 常温高湿(25 ℃/95%RH)、 甲苯和DMPA氛围储存后谐振频率的变化. 实验前将所有声学增强材料在80 ℃真空(-0.1 MPa)下加热2 h.
1.4.1 湿度对声学增强材料谐振频率的影响 在常温常湿或常温高湿储存实验中,实验持续时间为168 h. 将0.78 g分子筛型声学增强材料颗粒平铺在培养皿(直径10 cm)底部,培养皿敞开,分别放置在25 ℃/50%RH和25 ℃/95%RH的环境箱中,测试不同实验时间点材料的吸水量和谐振频率.
此外,将常温高湿环境下储存168 h 后的声学增强材料置于常温常湿环境下不同时间,测试声学增强材料的吸水量和谐振频率偏移值,以考察水分子吸附的可逆性及声学材料谐振频率的循环稳定性.
1.4.2 有机物小分子对声学增强材料谐振频率的影响 在有机物小分子氛围储存实验中,将0.40 g声学增强材料颗粒置于200 mL的广口瓶底部,将15 mL液态有机物置于25 mL广口瓶中,并将小广口瓶悬挂在大广口瓶盖上,密闭大广口瓶,用注射器反复多次抽除瓶内空气,在120 ℃的烘箱中放置4 h,测试声学增强材料的谐振频率及偏移值. 实验中采用的有机物小分子为甲苯和DMPA.
2 结果与讨论
2.1 声学增强材料的合成与结构
声学增强材料的合成路线如Scheme 1所示. 胶黏剂乳液与分子筛浆料(见本文支持信息)通过搅拌和匀浆除泡等操作,得到均一稳定的造粒母液. 将造粒母液通过喷雾造粒等方式制备成具有球形外观的声学增强颗粒材料. 最后,用不同目数的网筛进行筛分,选取粒径范围在300~350 µm的声学增强颗粒材料进行结构表征和谐振频率偏移值测试.

Scheme 1 Scheme of preparation of zeolitic acoustically enhanced materials
据文献[13]报道,对于以有机胺 (如TPA+)为模板剂合成的MFI分子筛,其拓扑对称性为正交晶系Pnma,每个单胞中存在12个结晶学不等价的骨架原子. 通过高温焙烧去除模板剂后,MFI分子筛结构的对称性转变为单斜晶系P21/n,每个单胞中具有24个结晶学不等价的骨架原子(表1),且正交和单斜晶系的MFI 分子筛结构在特定条件下互相转化,如吸附大体积的有机物分子、 改变外界温度或者压力[14],可以使单斜对称性的MFI分子筛结构转变为正交晶系的MFI分子筛结构.
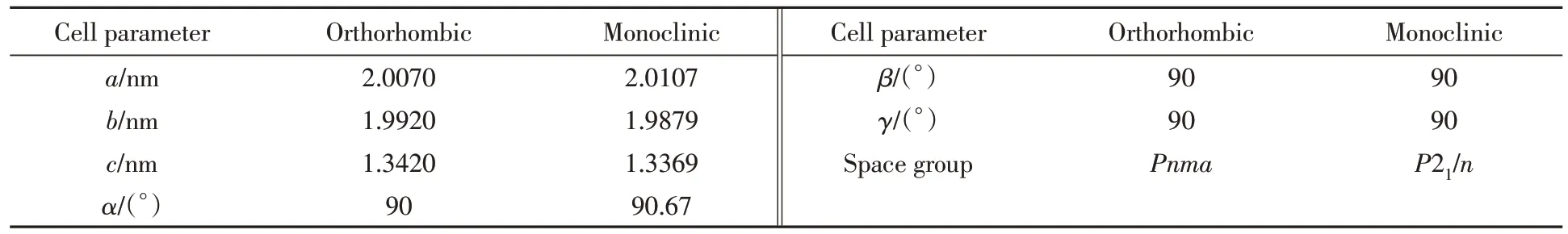
Table 1 Cell parameters of MFI under orthorhombic and monoclinic crystal systems[14]
在单斜晶系与正交晶系对称性的MFI分子筛结构发生转变的过程中,对应的XRD谱图在特定的衍射角可以观察到明显的区别,衍射角在24.4°,29.2°和48.6°的衍射峰出现单峰变为双峰或者单峰宽化的现象,表明由正交晶系转变为单斜晶系[15~19]. XRD 测试结果(图1)表明,不同硅铝比的分子筛均为MFI拓扑结构,但不同硅铝比的MFI分子筛对应的结构对称性有所不同. SAR=49.5(MFI_50)时,衍射谱图在24.4°和29.2°附近出现单峰,表明结构为正交晶系MFI分子筛拓扑结构; SAR>49.5时,衍射峰在24.4°和29.2°处发生劈裂或者宽化,具有明显的单斜晶系MFI 分子筛拓扑结构,与文献[16]报道一致. 检索国际分子筛协会结构委员会建立的分子筛结构数据库[20]可知,正交晶系和单斜晶系的MFI 分子筛结构均具有相同走向的二维交叉孔道,即[010]方向的十元环直孔道和[100]方向的十元环正弦孔道,二者交叉连通. 正交晶系MFI分子筛结构的十元环直孔道尺寸为0.522 nm×0.575 nm,十元环正弦孔道尺寸为0.529 nm×0.555 nm和0.528 nm×0.550 nm,而单斜晶系MFI分子筛结构的十元环直孔道尺寸为0.518 nm×0.578 nm 和0.527 nm×0.583 nm,十元环正弦孔道尺寸为0.535 nm×0.589 nm和0.501 nm×0.578 nm[21,22],更有利于气体的吸附和扩散.
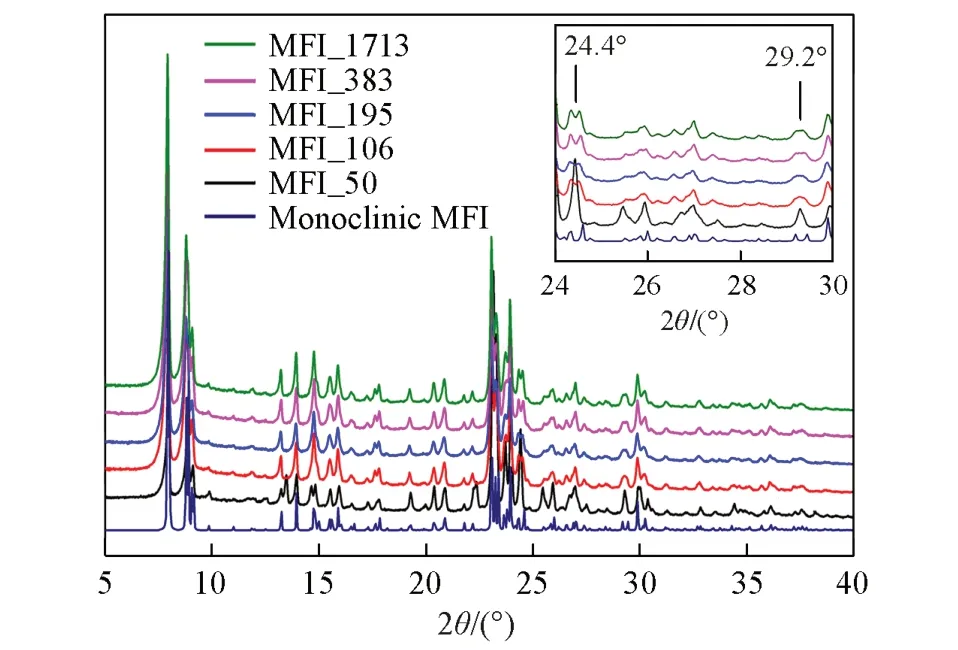
Fig.1 XRD patterns of zeolitic AEMs with various SARsInset: the evolution details of diffraction peaks between 24° and 30°.
对不同硅铝比沸石分子筛为主体材料的声学增强材料进行了低温(-196 ℃)氮气吸附测试. 测试前于120 ℃真空脱气2 h,低温氮气吸附等温线如图2所示. 随着硅铝比的升高,声学增强材料的比表面积增大,单层N2吸附量也增加. 当SAR<194.9时,在相对压力p/p0>0.45区域内存在介孔回滞环,SAR=105.7(MFI_106)时具有最大的介孔孔容; 当SAR>194.9时,声学增强材料的N2吸附-脱附曲线几乎完全重合,介孔孔容占比非常小,且具有相近的比表面积和微孔孔容.

Fig.2 N2 adsorption-desorption isotherms(-196 ℃) of zeolitic AEMs with various SARs(A) and pore size distribution of MFI_50(B), MFI_106(C) and MFI_195(D)“+10” implies the increasement with 10 cm3/g,STP from previous one.
不同硅铝比沸石分子筛的结构组成及低温N2吸附性能测试结果如表2所示. 为了排除骨架外阳离子种类对谐振频率的影响,实验中不同硅铝比分子筛的骨架外阳离子均为钠离子.

Table 2 Chemical compositions and textural properties of MFI zeolite with various SARs*
2.2 湿度影响
2.2.1 常湿储存实验 分子筛吸水通常有晶间和晶内吸水两种方式(图3). 对分子筛型声学增强材料在常温常湿条件下储存不同时间后吸水量的测试结果如图4所示. 可见,不同硅铝比的声学增强材料具有不同的初始吸水量. 声学增强材料的硅铝比越高,吸水量越低. 这主要是因为硅铝比越高,沸石分子筛结构的疏水性越强[23~25]. 在常温常湿条件下储存2 h后,SAR≥105.7的分子筛型声学增强材料的吸水量在很小范围内波动; 在常温常湿条件下储存3 h后,SAR=49.5的声学增强材料(MFI_50)的吸水量不再增加,达到饱和状态,其它硅铝比的声学增强材料的吸水量也几乎不变.

Fig.3 Illustration of hydration of zeolitic AEM
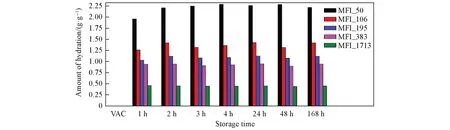
Fig.4 Amount of hydration of zeolitic AEM with various SARs after storage for different time at 25 ℃/50%RHVAC means storage in vacuum at 80 ℃ for 2 h.
对常温常湿条件下储存168 h 的声学增强材料进行了红外光谱测试. 如图5 所示,在3746 cm-1处存在明显的硅羟基(Si—OH)伸缩振动吸收峰; 3650~3700 cm-1的弱吸收峰为端位铝羟基(Al—OH)的伸缩振动吸收峰; 在3380~3400 cm-1范围内有宽且明显的吸收峰,对应水分子中羟基的伸缩振动;1630 cm-1附近的吸收峰为物理吸附的水分子的变形振动吸收峰; 1450~1550 cm-1范围内的吸收峰为分子筛中布朗斯特酸与路易斯酸的吸收峰; 1210 cm-1处的吸收峰为TO4四面体(T指代分子筛结构中的骨架原子,如Si,Al,P,B,Ga,Be等; TO4四面体为分子筛骨架元素与氧元素组成的最基本结构单元)外部连接T—O—T之间的反对称伸缩振动吸收峰; 1050 cm-1处的吸收峰为MFI分子筛中TO4四面体的反对称伸缩振动吸收峰; 780~800 cm-1处的吸收峰为MFI 分子筛中TO4四面体的对称伸缩振动吸收峰[26~30]. 超高硅结构(MFI_1713)虽具有一定的吸水量,但由于水含量很低及仪器检测限原因,红外谱图中几乎观察不到硅羟基、 铝羟基以及水分子羟基的振动峰. 随着硅铝比的增加,声学增强材料的吸水量降低,红外谱图中水分子对应的羟基振动峰逐渐减弱.
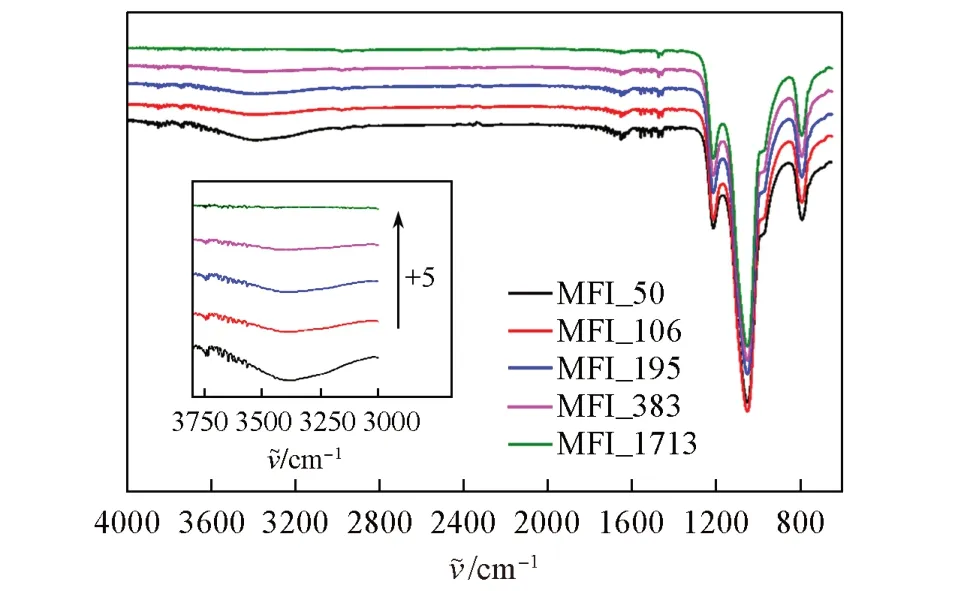
Fig.5 FTIR spectra of zeolitic AEMs with various SARs after storage at 25 ℃/50%RH for 168 hInset: zeolitic AEM’s transmittance between 3000 and 3800 cm-1; “+5” implies the increasement with 5%transmittance from previous one.
对常温常湿条件下储存168 h后的声学增强材料进行了XRD 测试,结果如图6所示. 在常温常湿条件下储存后,不同硅铝比的声学增强材料吸附了不同量的水分子(图4),但水分子的吸附并未改变分子筛结构的拓扑对称性(图6). 当SAR=49.5(MFI_50)时,2θ为24.4°和29.2°的单峰表明结构仍保持了正交晶系对称性. 当SAR>49.5 后,在24.4°和29.2°处的衍射峰发生劈裂或者宽化,表明结构具有单斜晶系对称性.
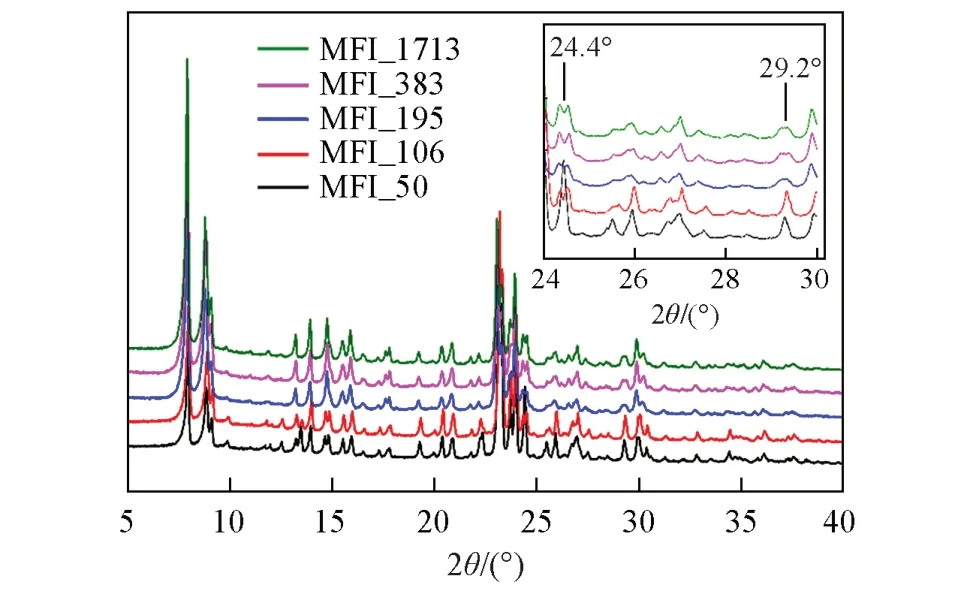
Fig.6 XRD patterns of zeolitic AEMs with various SARs after storage at 25 ℃/50%RH for 168 hInset: the evolution details of diffraction peaks between 24° and 30°.
随着硅铝比的升高,不同硅铝比分子筛为主体材料的声学增强材料的谐振频率偏移值(ΔF0,未加入与加入声学增强材料对应的谐振频率的差值)上升. 在常温常湿条件下储存1 h后,较低硅铝比(硅铝比≤105.7)声学增强材料的ΔF0明显降低; 储存2 h后,声学增强材料的吸水量达到饱和,ΔF0几乎不变(表S1,见本文支持信息),表明在水分子吸附饱和状态下,水分子的存在不影响声学增强材料的ΔF0. 在整个实验周期内,SAR≥194.9的声学增强材料的ΔF0出现微弱波动,说明常温常湿下水分子的存在并不影响材料的ΔF0(图7).
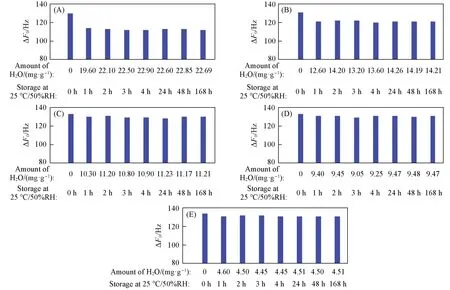
Fig.7 ΔF0 of zeolitic AEMs with various SARs stored for different time at 25 ℃/50%RH after storage in vacuum at 80 ℃ for 2 h(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
对常温常湿下储存168 h后的声学增强材料进行了常温(25 ℃)N2吸附性能测试,吸附-脱附等温线如图8所示. 测试结果表明,在经过常温常湿储存后,在25 ℃下不同硅铝比声学增强材料的N2吸附量随相对压力的增加近似呈线性增加,表明N2分子仍处于单层吸附状态. 在相同压力下,当SAR<200时,随着硅铝比的增加,常温N2吸附量增加; 当SAR>200 时,常温N2吸附量在微小范围内波动. 与低温N2吸附结果(表2)相比,在相对压力p/p0=1.0 时,常温N2吸附量非常低(表3),硅铝比为383.0(MFI_383)的声学增强材料具有最大的常温N2吸附量,为6.2827 cm3/g,STP,表明常温吸附的N2分子占据了非常少的吸附位点或比表面积.
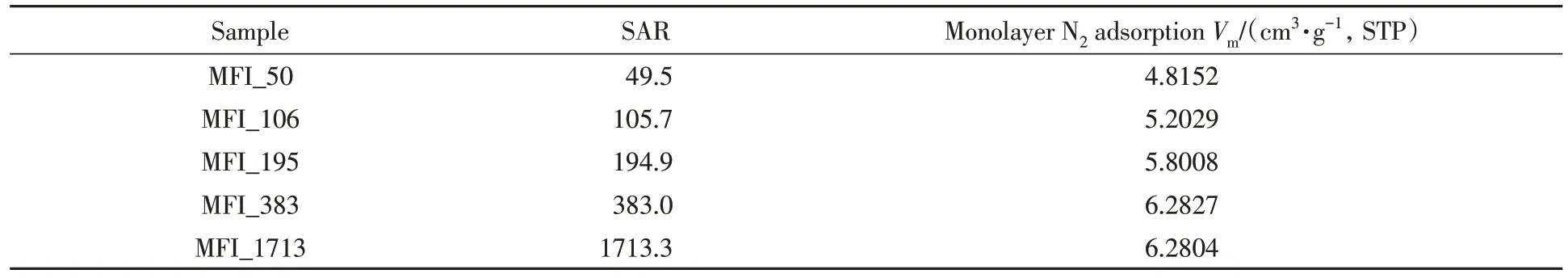
Table 3 Monolayer N2 adsorption(25 ℃) of zeolitic AEMs after 168 h storage under 25 ℃/50%RH
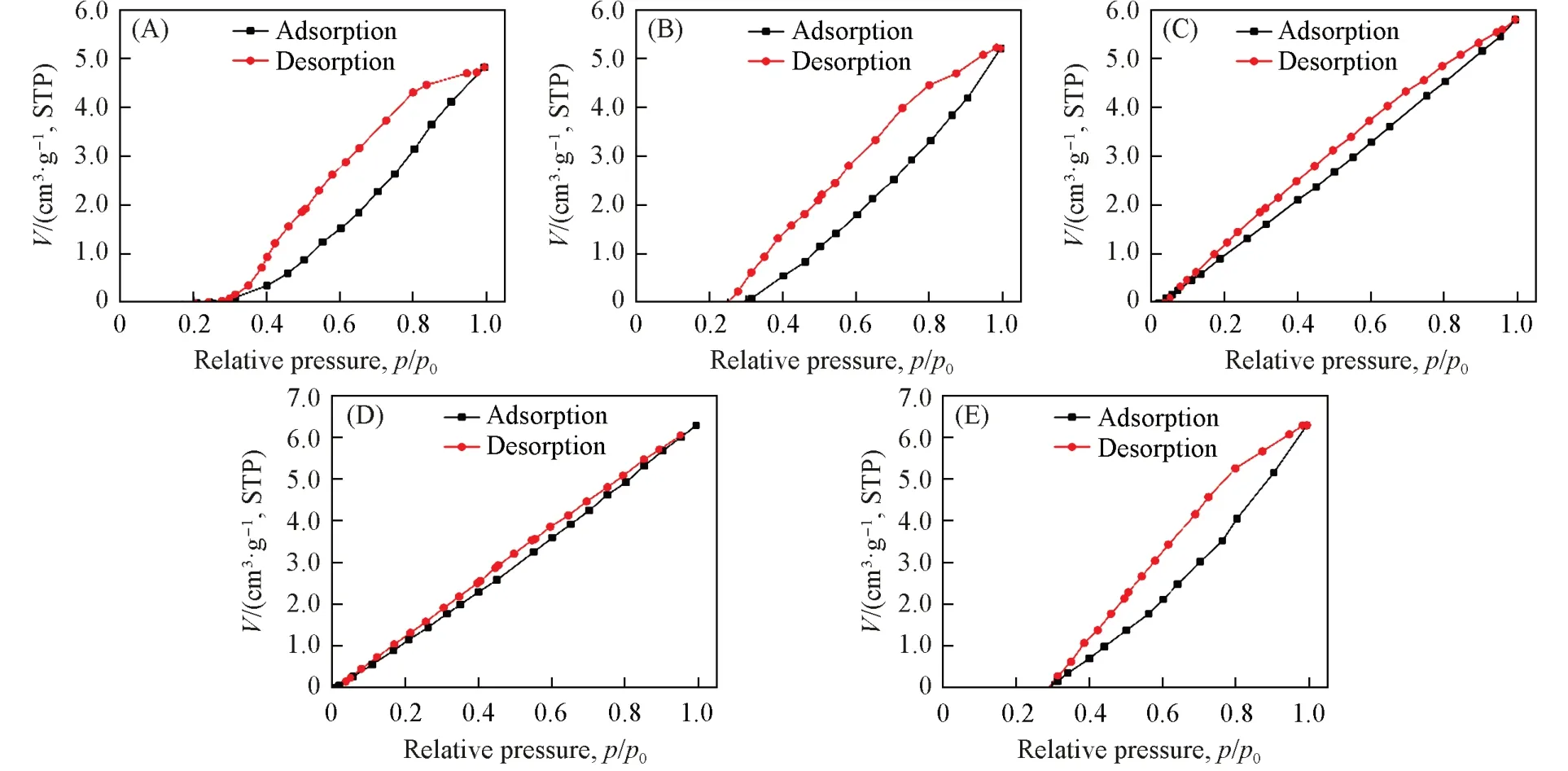
Fig.8 N2 adsorption-desorption isotherms(25 ℃) of zeolitic AEMs after 168 h storage at 25 ℃/50%RH(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
2.2.2 高湿储存实验 不同硅铝比的声学增强材料在常温高湿条件下储存后的吸水量测试结果如图9所示. 在常温高湿条件下储存2 h后,低硅铝比(MFI_50)的声学增强材料吸水量快速增加并达到饱和,超高硅结构的声学增强材料(MFI_1713)因饱和吸水量相对较低,在常温高湿下储存2 h后,几乎达到饱和. 在常温高湿下储存3 h后,所有不同硅铝比的声学增强材料的吸水量几乎不再发生变化,说明不同硅铝比的声学增强材料的吸水量达到饱和.
对常温高湿下储存168 h 的声学增强材料进行了红外光谱测试. 如图10 所示,由于水分子的存在,红外谱图中出现羟基伸缩振动峰(3380~3400 cm-1); 随着硅铝比的降低,声学增强材料的吸水量升高,在红外谱图上可以观察到水分子对应的羟基伸缩振动峰逐渐增强. 常温高湿下储存后,由于吸水量较常温常湿下储存有明显的增加,红外谱图中水分子对应的羟基伸缩振动峰更加明显.
“宝剑锋从磨砺出”。面对纷繁复杂的食品安全形势和变化多端的违法犯罪行为,哈斯巴彦尔不顾个人安危,勇挑重担,身先士卒,在全区各地,留下了拼搏的身影和跋涉的足迹。2014年以来,他克服执法人员少、队伍新、任务重等困难,带领同志们主动出击,查办多起在全区有影响的食品类大案要案,涉案货值金额2亿多元。执法过程中,他多次受到威胁,但从未退缩。

Fig.10 FTIR spectra of zeolitic AEMs with various SARs after 168 h storage at 25 ℃/95%RHInset: zeolitic AEM’s transmittance between 2750 and 4000 cm-1.
对常温高湿下储存168 h 的声学增强材料进行了XRD 测试,结果如图11 所示. 当SAR=49.5(MFI_50)时,24.4°和29.2°处的单峰表明结构仍保持了正交晶系对称性. 与常温常湿条件下的储存结果相比,SAR=105.7(MFI_106)时,24.4°处的峰形发生微弱劈裂,但29.2°处对应的衍射峰仍保持宽化,表明结构具有明显的单斜晶系对称性; SAR>105.7 后,声学增强材料的结构对称性未出现变化.以上结果表明,常温高湿下水分子的吸附量不足以改变声学增强材料中分子筛的拓扑对称性.

Fig.11 XRD patterns of zeolitic AEM with various SARs after 168 h storage at 25 ℃/95%RHInset: the evolution details of diffraction peaks between 24° and 30°.
在常温高湿储存过程中,以不同硅铝比沸石分子筛为主体材料的声学增强材料的ΔF0值(表S2,见本文支持信息)随着硅铝比的升高而增大. 在常温高湿下储存1 h后,较低硅铝比(≤105.7)的声学增强材料的ΔF0出现明显的降低,但随着实验时间超过2 h,声学增强材料的吸水量趋于饱和,ΔF0维持在较小的范围内波动,说明吸水饱和后ΔF0维持稳定. 对于高硅铝比(194.9~383.0)的声学增强材料,在实验时间超过2 h后,声学增强材料的吸水量仍有所增加,但对应的ΔF0波动微弱,未发生明显偏移. 在整个实验周期内,随着吸水量的增加,超高硅结构声学增强材料(MFI_1713)的ΔF0几乎未发生变化(图12). 以上结果表明,硅铝组成的声学增强材料在常温高湿下吸水饱和后,水分子的存在不影响ΔF0,超高硅组成的声学增强材料的ΔF0受吸水量影响很小.

Fig.12 ΔF0 of zeolitic AEM with various SARs stored for different time at 25 ℃/95%RH after storage in vacuum at 80 ℃ for 2 h(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
对常温高湿储存168 h后的声学增强材料进行了常温(25 ℃)N2吸附性能测试,N2吸附-脱附等温线如图13所示. 测试结果表明,在经过常温高湿储存后,在25 ℃下不同硅铝比声学增强材料的N2吸附量随相对压力的增加近似呈线性增加,表明N2分子仍处于单层吸附状态. 在相对压力p/p0=1.0时,吸附量如表4所示. 在相同压力下,当SAR<200时,随着SAR的增加,常温N2吸附量增加; 当SAR>200时,常温N2吸附量在微小范围内波动. 与低温N2吸附结果(表2)相比,在相对压力p/p0=1.0时,常温N2吸附量非常低. SAR=383.0 的声学增强材料(MFI_383)具有最大的常温N2吸附量,为6.0360 cm3/g,STP,表明常温下吸附的N2分子只占据了非常少的吸附位点或比表面积. 与表3中常温常湿储存后的N2吸附结果相比,由于高湿环境中水分子的吸附量增加,水分子占据了部分最优吸附位点,导致N2吸附量降低,对应硅铝比的声学增强材料的ΔF0微弱减小.
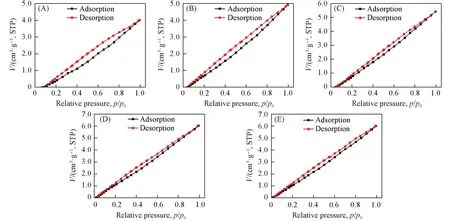
Fig.13 N2 adsorption-desorption isotherms(25 ℃) of zeolitic AEMs after 168 h storage at 25 ℃/95%RH(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
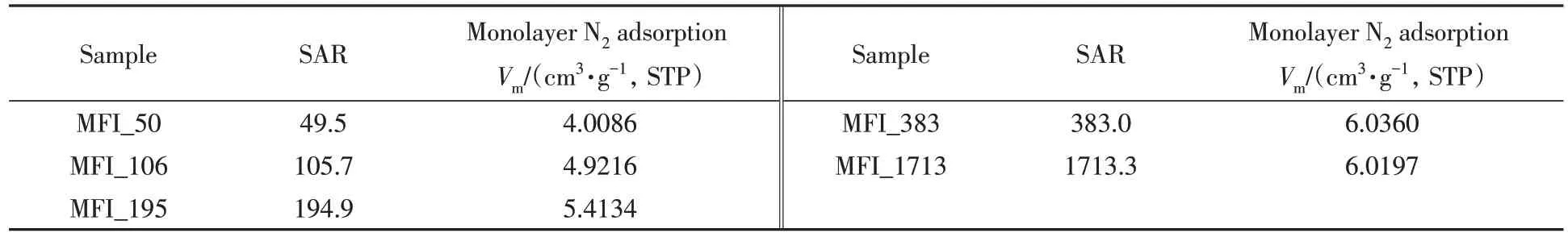
Table 4 Monolayer N2 adsorption(25 ℃) of zeolitic AEM after 168 h storage at 25 ℃/95%RH
在常温高湿实验后,将分子筛型声学增强材料在常温常湿条件(25 ℃/50%RH)静置不同时间,观察声学增强材料中水分的变化. 随着静置时间增长,材料在高湿情况下吸附的水分子逐步脱附. 静置时间超过48 h后,水分子脱附与吸附达到平衡状态,吸水量保持恒定(图14).
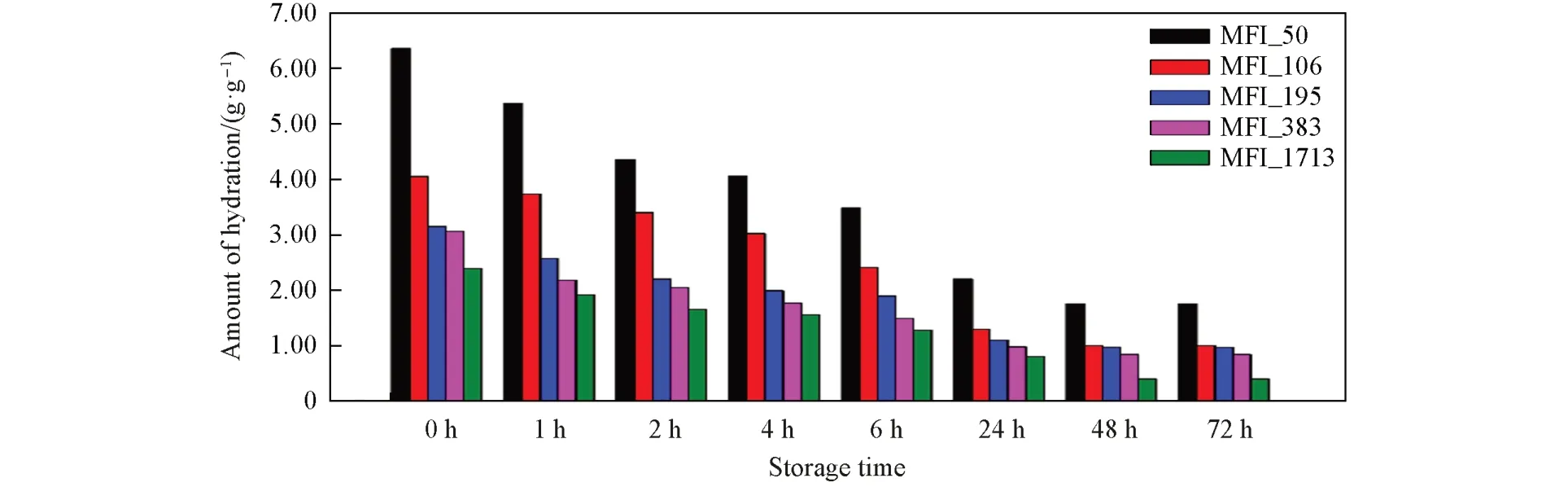
Fig.14 Amount of hydration of zeolitic AEMs with various SARs stored for different time at 25 ℃/50%RH after 168 h storage at 25 ℃/95%RH
常温高湿储存实验结束后,将不同硅铝比分子筛为主体材料的声学增强材料于常温常湿环境中静置不同时间. 在常温常湿下静置1 h后,低硅铝比的声学增强材料(MFI_50)的ΔF0明显恢复,但随着实验时间延长,ΔF0维持在较小的范围内波动. 当常温常湿静置时间>1 h后,高硅铝比(105.7~383.0)的声学增强材料的ΔF0存在恢复现象. 在整个实验周期内,超高硅结构声学增强材料(MFI_1713)的ΔF0几乎没有变化(图15). 不同硅铝比的声学增强材料在常温常湿环境中静置4 h后,与常温常湿储存的ΔF0几乎相同(表S3,见本文支持信息),表明水分子的吸附可逆. 结合后处理不同时间材料的水吸附量,发现在后处理4 h后,吸水量持续降低,但对应的ΔF0几乎不变,表明常温下水分子的吸附不影响声学增强材料的ΔF0.

Fig.15 ΔF0 of zeolitic AEM with various SARs stored for different time at 25 ℃/50%RH after 168 h storage at 25 ℃/95%RH(A) MFI_50;( B) MFI_106;( C) MFI_195;( D) MFI_383;( E) MFI_1713.
2.3 有机物小分子的影响
在有机物小分子氛围下储存相同时间后,不同硅铝比声学增强材料的ΔF0随着硅铝比的升高而降低. 以甲苯作为有机物小分子的储存实验过程中,较低硅铝比(SAR≤105.7)声学增强材料的ΔF0≥100 Hz. 在储存1 h后,SAR≥194.9的声学增强材料具有非常接近的ΔF0(≥110 Hz,图16). 在甲苯氛围中储存1 h后,不同硅铝比的声学增强材料的ΔF0均出现明显的降低,但实验时间超过2 h后,ΔF0维持在较小的范围内波动(表S4,见本文支持信息). 甲苯分子是一种弱极性有机物分子,其甲基在横向方向与苯分子尺寸几乎一致,约为0.53 nm[31],与MFI拓扑结构中的孔道尺寸接近[32],MFI分子筛的拓扑结构中有[001]方向的十元环正弦孔道和[010]方向的十元环直孔道[33]. 因此,处于吸附状态的甲苯分子在取向与直孔道走向平行时,可自由进行吸附-脱附,处于正弦孔道时,由于分子取向与孔道走向存在偏差,吸附状态的甲苯分子在脱附扩散过程中会受到一定限制.

Fig.16 ΔF0 of zeolitic AEMs with various SARs stored for different time in volatile toluene after storage in vacuum at 80 ℃ for 2 h(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
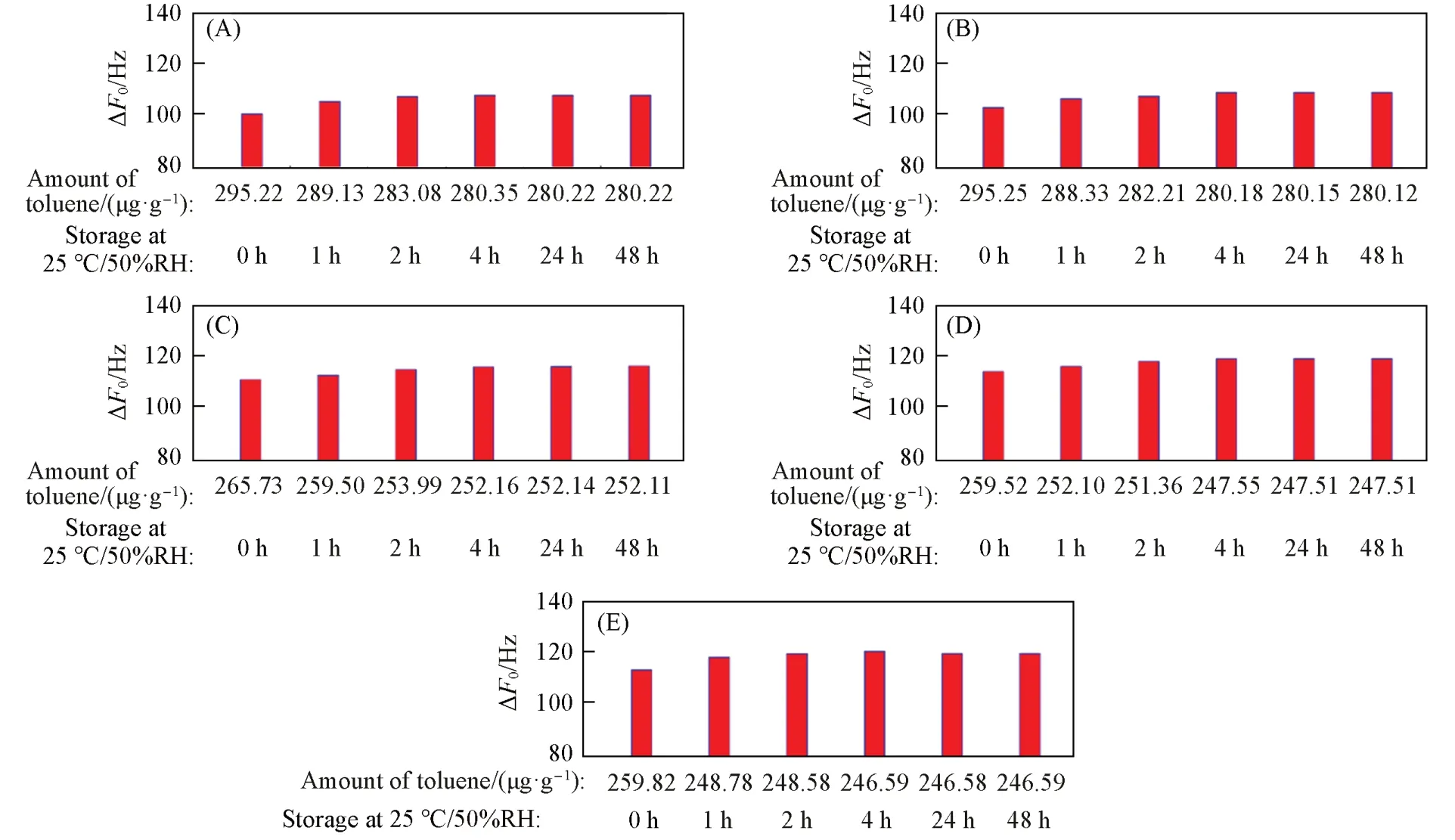
Fig.17 ΔF0 of zeolitic AEMs with various SARs stored for different time at 25 ℃/50%RH after 24 h storage in volatile toluene(A) MFI_50;( B) MFI_106;( C) MFI_195;( D) MFI_383;( E) MFI_1713.
在有机物小分子DMPA 氛围中储存24 h后,超高硅结构声学增强材料(MFI_1713)的ΔF0接近110 Hz,高硅铝比(SAR=194.9,383.0)声学增强材料的ΔF0介于85~95 Hz之间,SAR≤105.7的声学增强材料ΔF0明显降低,为60~70 Hz(表S6,见本文支持信息). 在DMPA氛围中储存1 h后,不同硅铝比的声学增强材料的ΔF0出现明显的降低,实验时间超过2 h 后,ΔF0几乎不变(图18). DMPA 是一种强极性有机物分子,其结构中的甲基和碳碳双键的尺寸均小于MFI拓扑结构孔道,可自由进入孔道中,但由于DMPA为非线性结构,因此,DMPA分子整体进入孔道的概率很小. 结构中的羰基、 碳碳双键及氮原子均可与分子筛中的铝原子、 羟基基团和骨架外阳离子进行作用,形成嵌入式吸附[34]. 随着硅铝比的升高,结构中的铝原子和骨架外阳离子减少,可形成嵌入式吸附的位点减少,DMPA分子吸附量减少,提高了氮气分子的进出几率.
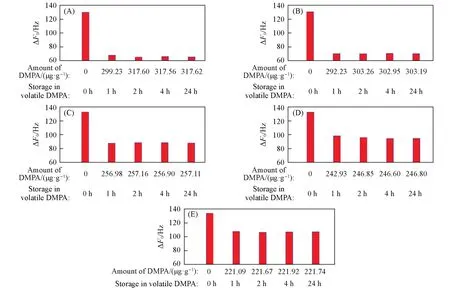
Fig.18 ΔF0 of zeolitic AEM with various SARs stored for different time in volatile DMPA after storage in vacuum at 80 ℃ for 2 h(A) MFI_50; (B) MFI_106; (C) MFI_195; (D) MFI_383; (E) MFI_1713.
将在DMPA氛围中储存24 h后的声学增强材料于常温常湿(25 ℃/50%RH)下放置不同时间,测试了DMPA残余量和对应的ΔF0,结果如图19所示. 不同硅铝比的声学增强材料DMPA残余量降低十分微弱,ΔF0变化微弱(表S7,见本文支持信息).
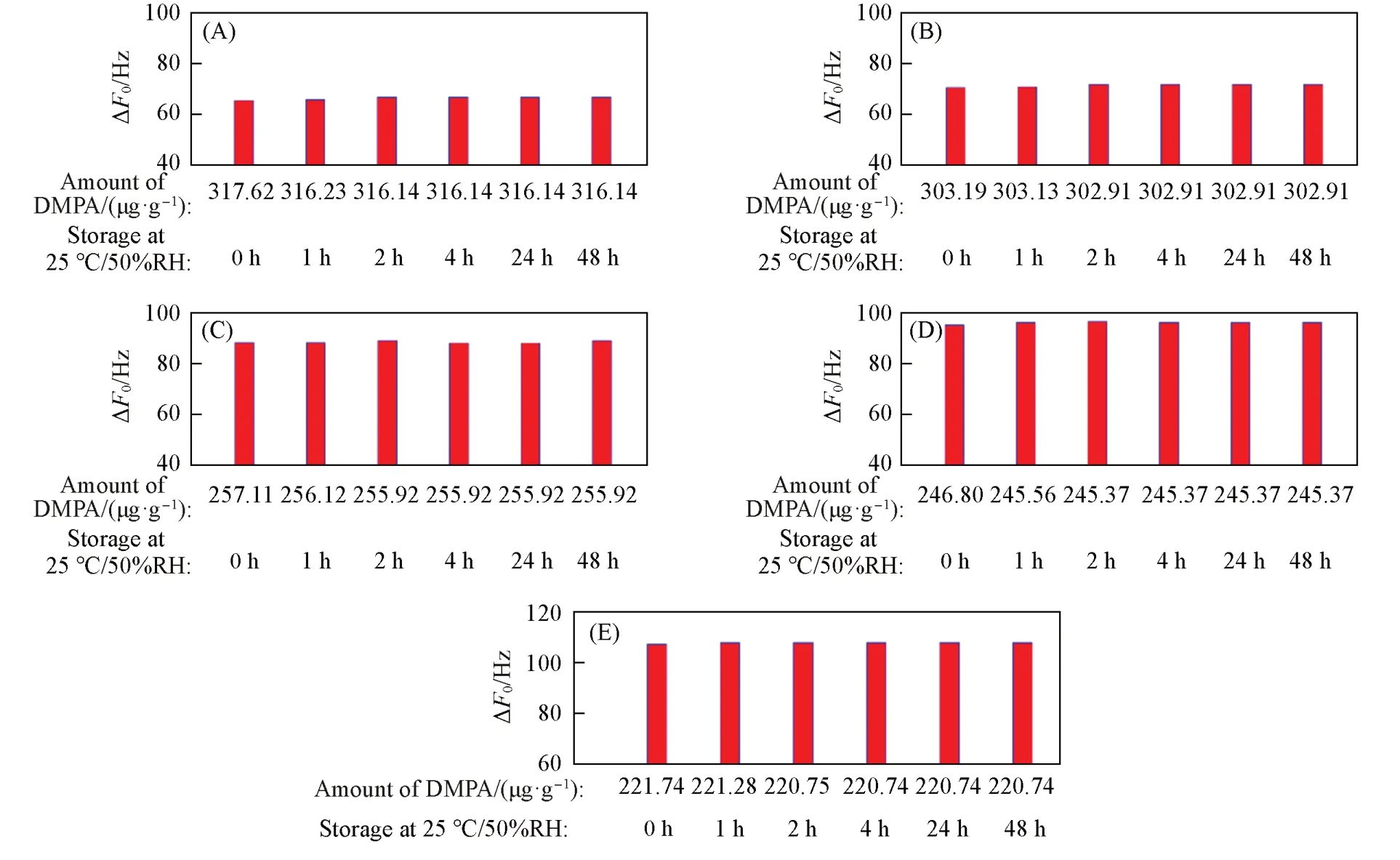
Fig.19 ΔF0 of zeolitic AEM with various SARs stored for different time at 25 ℃/50%RH after 24 h storage in volatile DMPA(A) MFI_50;( B) MFI_106;( C) MFI_195;( D) MFI_383;( E) MFI_1713.
据文献[35~37]报道,MFI拓扑结构在吸附有机物或存在较多水合阳离子时,拓扑对称性会由单斜晶系向正交晶系转变,结构中的铝含量也会影响拓扑对称性的转变. 在甲苯氛围中储存后的声学增强材料XRD测试结果如图20(A)所示. 当SAR=49.5(MFI_50)时,在2θ为24.4°和29.2°处的单峰表明结构仍保持了正交晶系对称性,说明甲苯的吸附不影响结构对称性. 当 SAR=105.7(MFI_106)时,在24.4°和29.2°两处的衍射峰由常温常湿状态下的双峰或宽峰变为单峰,说明结构对称性由单斜晶系转变为正交晶系,甲苯的吸附改变了SAR=105.7(MFI_106)的分子筛结构对称性. 当SAR>105.7 时,声学增强材料中的分子筛结构对称性仍保持单斜晶系,说明甲苯吸附量较少不足以改变结构对称性.
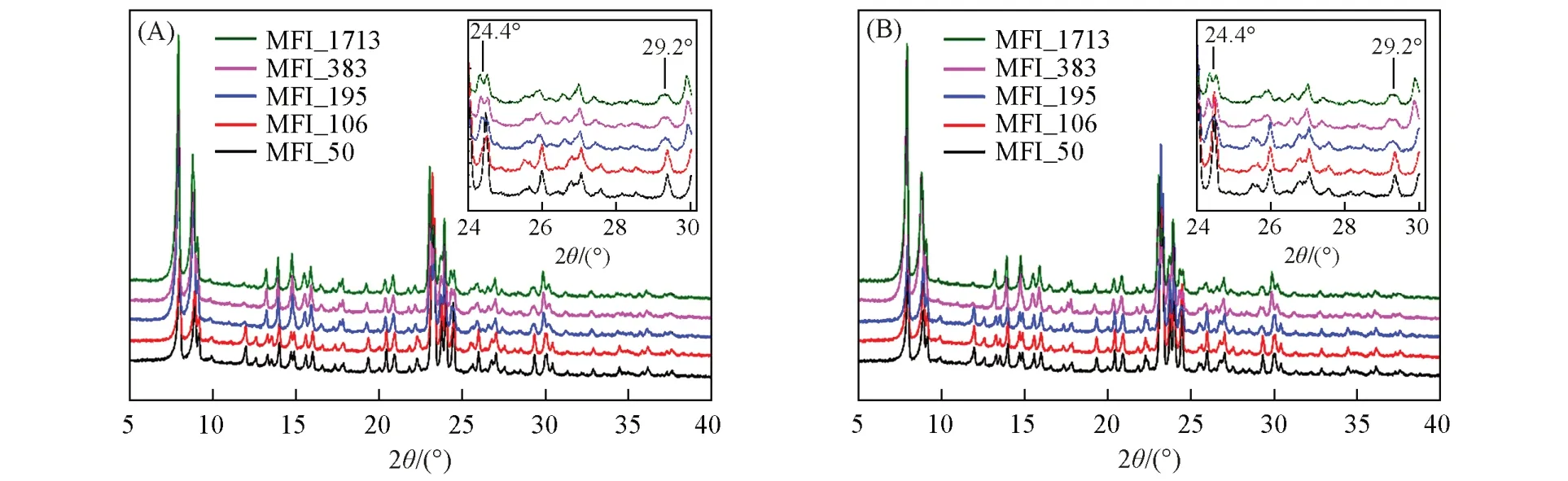
Fig.20 XRD patterns of zeolitic AEMs with various SARs after 24 h storage in volatile toluene(A)and DMPA(B)Insets: the evolution details of diffraction peaks between 24° and 30°.
声学增强材料在DMPA 气氛中储存后,SAR=105.7(MFI_106)时,在24.4°和29.2°处的衍射峰由常温常湿状态下的双峰或宽峰变为单峰,表明结构对称性由单斜晶系转变为正交晶系. SAR=194.9(MFI_195)时,24.4°处的衍射峰由常温常湿状态下的双峰变为较宽的单峰,29.2°处的衍射峰由常温常湿状态下的宽峰变为馒头峰,结构对称性发生改变. 在SAR>194.9后,声学增强材料的中分子筛的结构对称性仍保持单斜晶系,表明DMPA吸附量较少不足以改变结构对称性[图20(B)].
对在甲苯氛围中储存24 h的声学增强材料进行了红外光谱测试,结果如图21(A)所示. 不同硅铝比的声学增强材料在甲苯氛围中储存后,均可观察到甲苯的特征吸收峰. 在3030~3080 cm-1范围内的吸收峰为苯环中C—H伸缩振动吸收峰; 2960和2870 cm-1附近的吸收峰为甲基中C—H伸缩振动吸收峰; 1604和1498 cm-1附近的吸收峰为苯环骨架振动吸收峰. 对在DMPA氛围中储存24 h的声学增强材料进行了红外光谱测试,结果如图21(B)所示. 不同硅铝比的声学增强材料在DMPA 氛围中储存后,均可以观察到DMPA 的特征吸收峰. 2940 cm-1附近的吸收峰为碳碳双键中碳的C—H 伸缩振动吸收峰; 1646 cm-1处的吸收峰为羰基的伸缩振动吸收峰; 1607 cm-1处的吸收峰为碳碳双键的振动吸收峰;1417 cm-1处的吸收峰为氮碳键的伸缩振动吸收峰.

Fig.21 FTIR spectra of zeolitic AEMs with various SARs after 24 h storage in volatile toluene(A)and DMPA(B)Insets zeolitic AEMs’ transmittance between 2500 and 4000 cm-1. “+5” implies the increasement with 5%transmittance from previous one.
3 结 论
研究了不同湿度和不同有机物小分子对不同硅铝比分子筛组成的声学增强材料声学性能的影响.与低温氮气吸附结果相比,常温下声学增强材料对氮气的吸附量很低,即使具有较高吸水量的低硅铝比沸石分子筛结构中仍存在足够的氮气分子吸附位点. 因此,常温下氮气分子的吸附量不会因吸水量的变化而发生明显变化,声学增强材料的ΔF0不会受到明显影响. 不同种类的有机物小分子在声学增强材料中的吸附状态有所不同,分子动力学直径与沸石分子筛孔径相似或略小的有机物小分子可在一定程度上进行可逆的吸附-脱附,对声学增强材料的ΔF0影响较小. 分子动力学直径大于沸石分子筛孔径或具有小尺寸端基基团的有机物小分子,会在声学增强材料中形成嵌入式吸附,降低对氮气分子的吸附,从而影响声学增强材料的ΔF0. 本研究结果可为开发更优异的声学增强材料提供借鉴.
支持信息见http: //www.cjcu.jlu.edu.cn/CN/10.7503/20230474.
猜你喜欢
钻井液与完井液(2022年4期)2022-10-26
数学物理学报(2022年4期)2022-08-22
中学生数理化·中考版(2021年10期)2021-11-22
昆明医科大学学报(2021年8期)2021-08-13
哈尔滨轴承(2021年1期)2021-07-21
家庭影院技术(2020年6期)2020-07-27
家庭影院技术(2019年1期)2019-01-21
家庭影院技术(2018年11期)2019-01-21
家庭影院技术(2018年10期)2018-11-02
电镀与环保(2017年3期)2017-06-23
