
不同氮源用于电催化合成氨的研究进展
2024-03-25 02:42:16赵晓光王云龙尹海波曲亚坤苏海伟
高等学校化学学报 2024年3期
赵晓光,王云龙,尹海波,曲亚坤,苏海伟,房 韡
(1. 中石化石油化工科学研究院有限公司, 北京 100083;2. 清华大学环境学院, 北京 100084)
氨(NH3)是化肥、 制药和化工行业中的重要原料,NH3分子因具有高能量密度(3.5 kW·h/L)和高氢质量分数(17.5%)的优点被视为理想的无碳储能燃料[1~5],并且NH3在储存和应用方面的成本显著低于氢气(H2). 通过氮气(N2)和H2发生还原反应(Nitrogen reduction reaction,NRR)合成NH3的Haber-Bosch工艺是20 世纪最具有影响力的发明之一,该发明贡献了全球NH3产量的90%,每年产量约为1.75 亿吨,其化学反应主要如下[6]:
在该放热反应中,高压和低温的条件更利于NH3的合成. 但在动力学角度上,适当提高反应温度有利于提高反应速率、 加快工业生产的进程. 然而,NH3稳定性会因温度的升高而变弱,降低产品的收率,所以需要进一步提高反应压力以抑制产物NH3的热分解. 同时,考虑到需断裂高惰性的N≡N 键(941.82 kJ/mol)[7],Haber-Bosch 工艺的应用需要提供300~500 ℃的高温和100~300 atm(1 atm=1.013×105Pa)高压条件,造成每年消耗的能源约占全球总能源消耗的1%,产生的二氧化碳(CO2)超过全球CO2总排放量的1.4%[8,9]. 其中,反应所需的高纯H2是对天然气等化石燃料进行催化重整流程制备的,制备与纯化步骤繁琐、 能耗高[10]. Haber-Bosch工艺在合成NH3过程中对于资源的大量消耗加剧了全球变暖和环境污染,因此,人们在探索温和条件下的NH3合成替代途径方面做出了巨大努力,开发了酶催化[11]、 电催化[3,12]、 光催化[13,14]、 等离子体驱动转化[15~17]等方法. 电催化合成NH3方法不受热力学限制,可以在常温常压的温和条件下进行,反应过程绿色环保,相比Haber-Bosch工艺至少能够减少20%的能源消耗[3],已成为近年来的研究热点. 且与在大型工厂集中生产NH3的Haber-Bosch工艺相比,电催化合成NH3可以在小型设备中实现,能够支持分布式NH3与化肥生产[10,18]. 但电催化合成NH3仍存在诸多挑战,如NH3生成的速率和法拉第效率低等. 如何提高电催化合成NH3的性能对于实现NH3的工业化生产至关重要. 电催化合成NH3反应一般选用含有电解质的水溶液作为电解液,导致电催化合成氨性能容易受到析氢反应(Hydrogen evolution reaction,HER)副产物的竞争影响. 此外,使用N2、 硝酸根和一氧化氮(NO)作为不同氮(N)源用于电催化合成NH3反应时存在不同的问题,如N2难以溶解于水被吸附与活化、还原因N元素价态跨度大而难以控制反应路径和NO体系复杂等,阻碍了高效高稳定性电催化合成NH3. 解决以上问题的关键在于根据不同反应N源优化相应的反应装置、 开发高活性高稳定性的催化剂、 选用不同的电解质体系以提高电催化合成NH3的效率和稳定性. 为此,本文从不同N 源用于电催化合成NH3的反应机理出发,介绍了构建高效高稳定性合成NH3体系的方法,总结了提高催化剂活性和稳定性的设计策略和思路,综合评述了利用不同N源电催化合成NH3的催化剂的研究进展,并展望了该领域存在的潜在挑战和应用前景.
1 以N2为N源
1.1 电催化NRR合成NH3机理
N是人类社会和自然界不可缺少的元素,在自然界中最丰富的N源是大气中约占78%的N2[19]然而,N2分子因极低电子亲和能(-1.8 eV)、 高解离能(410 kJ/mol)和最高占据分子轨道-最低未占分子轨道(HOMO-LUMO)能隙(22.9 eV),在温和条件下很难实现NH3合成的反应[20~22]. 新兴的电催化NRR能够在温和的条件下利用可再生能源合成NH3,展现了巨大的发展潜力和应用价值. 与传统Haber-Bosch工艺NRR 相比,电催化NRR 合成NH3具有诸多优点[18,23,24]: (1) 在电化学反应器中合成NH3能够突破反应热力学限制,实现温和条件下的N2分子活化与转化,避免耗费大量的热能; (2) 该工艺简便易操作,可利用可再生能源发电进行小规模、 分散式制NH3; (3) 选用水而非H2作为电化学合成NH3的氢源,避免使用化石燃料制氢造成能量消耗和CO2排放.
电催化NRR合成NH3在阴极发生还原反应,主要包括以下步骤[25]: (1) N2溶解在水中被吸附于催化剂表面; (2) N2通过解离、 缔合或晶格氧(Mars-Van Krevelen)机理3 种反应机制结合H 原子被还原(图1)[3]; (3) 合成的NH3于催化剂表面脱附. 在解离机制中,吸附状态N2分子的N≡N键被破坏后,两个独立的N原子同时发生氢化反应,最终分别生成NH3并于催化剂表面脱附. 在缔合机制中,N2分子吸附于催化剂表面,两个N原子始终保持化学键连接,最后N—N键断裂生成NH3分子. 缔合机制又分为交替配位和末端配位两种方式: 在交替配位方式中,催化剂表面的两个N 原子交替进行加氢步骤,两个NH3分子先后脱去; 在末端配位方式中,远离催化剂表面的N原子上首先完成加氢后发生N—N键断裂脱去一分子NH3,随后内侧N原子氢化,形成一分子NH3后自表面脱附. Mars-Van Krevelen机制通常发生在过渡金属氮化物表面,过渡金属氮化物上的N首先被还原为NH3后,N2分子填补到N空位处继续发生还原反应生成NH3,该机制能够有效加速传质动力学过程[26].

Fig.1 Mechanism of electrocatalytic NRR for NH3 synthesis
1.2 电催化NRR合成NH3性能调控
电催化合成NH3性能(活性与选择性)主要由产生NH3的速率与法拉第效率进行评价[27]. 由于反应体系限制、 竞争副反应的存在与催化剂结构仍需调控,电催化NRR 合成NH3效率远未达到商业化标准[28]. 其中存在的主要技术难题为: (1) 反应原料N2溶解度低(0.66 mmol/L),其传质与吸附过程的限制影响了整体NRR 的催化活性[29]; (2) N≡N 键的高键能使其断裂极为困难,N2的活化成为电催化NRR 的决速步骤,降低了电催化NRR 合成NH3的产率[30,31]; (3) 选用水(H2O)作氢源不可避免会发生HER,HER 与NRR 电位相近,且H2O 更易被活化产生质子,因此,HER 在动力学上更易在阴极发生,大大降低NH3合成的法拉第效率[32,33]; (4) NH3分子合成后需及时从催化剂表面脱附,若催化剂吸附能力过强会导致产物NH3无法脱附,若吸附能力过弱则无法有效进一步活化N2. 针对以上问题,目前的研究主要集中在对催化剂材料进行调控与设计和对电催化反应系统进行改进和优化[13,32,34,35].
科研人员首先通过密度泛函理论(Density functional theory,DFT)计算出不同金属表面NRR活性的“火山型”曲线以高效设计NRR 电催化剂(图2)[36],发现钌(Ru)、 钼(Mo)、 铁(Fe)和铑(Rh)可以表现出最低电位下的NRR高催化活性,但其位于灰色区域,吸附H的能力高于吸附N 的能力,HER 竞争反应较NRR剧烈,需调控HER 和NRR 速率以提高合成NH3效率. 同时前过渡金属(未着色区域钪、 钇、 钛和锆)表面吸附N 的能力强于吸附H 的能力,也有望成为有前途的NRR 电催化剂. 根据NRR 电催化剂的组成和特性不同,将其分为贵金属基催化剂、 非贵金属基催化剂和非金属基催化剂3类.
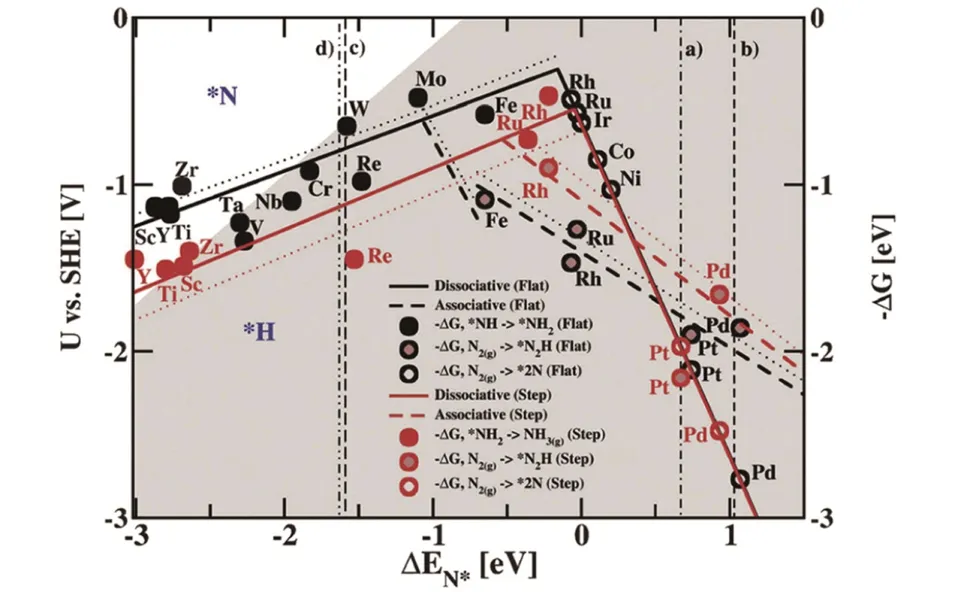
Fig.2 “Volcano” diagram for theoretical calculation of NRR activities on metal surfaces[36]Copyright 2012,the Royal Society of Chemistry.
1.2.1 贵金属催化剂 贵金属催化剂因表面极强的结合能和优异的导电能力被广泛应用于电催化反应中. 如Ru 原子外层拥有一个空d轨道,能够接受N2分子提供的孤电子用于活化N≡N 键,提高合成NH3的效率[37]. 以石墨为载体的Ru/C电催化剂作为NRR的阴极已早有报道[38],但其表现出较低的NH3产率(0.21 µg·h-1·cm-2)和法拉第效率(0.28%). Back等[39]选择Ru金属作为模型探究N≡N键的解离步骤,发现Ru表面的NRR高选择性可能是由于其对关键中间体*HNNH吸附较强促使NH3的形成和副产物*NH2NH2的减少,但Ru 在低电位下会因HER 活性较高使得H 原子占据NRR 的活性位点,提高了N2的活化能垒,降低了反应效率. 通过调控Ru金属的存在形式和选择合适的载体,能够进一步改善NRR合成NH3的性能. 如,Liu 等[34]将Ru 单原子分散于二氧化锆(ZrO2)表面,Ru 单原子与ZrO2的氧空位(Oxygen vacancies,Vo)协同促进作用有效抑制了HER,从而提高了NH3合成的法拉第效率(21%). DFT计算表明,该催化剂在低电位下能够稳定吸附N2以生成*NNH,削弱HER副反应的竞争,具有较高的催化活性. 金(Au)原子外层的空d轨道同样能够接受N2的孤电子对,并且与Ru相比具有较低的HER活性. Bao等[40]制备了具有被(210)和(310)晶面包围的阶梯式的(730)晶面的二十四面体Au纳米棒,该阶梯面上Au原子处于不饱和配位状态,作为活性位吸附N2分子形成Au—N键,以缔合机制的交替配位方式加氢高效合成NH3. Hu等[41]探索了Au表面独特的等离子共振效应对电催化NRR的影响,将Ru纳米簇覆盖于Au纳米簇上,Au表面的等离子共振效应能够产生局域电场,加速热电子在Ru表面的转移以加速N2活化. 此外,Rh和钯(Pd)也在电催化NRR合成NH3中得到应用. Liu等[42]构建了超薄Rh纳米片电催化剂,Rh的三维互联结构和低配位的原子缺陷有助于提高NRR合成NH3的活性. Yao等[43]采用在线微分电化学质谱(Differential electrochemical mass spectrometry,DEMS)[图3(A)]和表面增强红外吸收光谱[图3(B)]手段检测反应中间体,以研究Rh金属表面电催化NRR合成NH3的反应路径,提出Rh 表面的NRR 合成NH3过程可能分为N2还原为N2H2过程和在电解液中的分解过程. Wang 等[44]将Pd 负载于炭黑基底(Pd/C),在磷酸盐缓冲盐溶液(Phosphate buffer saline,PBS)中具有高NH3产率(约4.5 µg·mg-1·h-1)与高法拉第效率(8.2%),通过密度泛函理论计算表明,Pd 独特的NRR 合成NH3活性来源于对HER反应的抑制与质子跳跃(Grotthuss)机制,降低了加氢反应的能量势垒.

Fig.3 DEMS of intermediates on Rh/C during a scan in a N2-saturated 1 mol/L KOH solution(A)and FTIR spectra during the first segment from 0.4 V to -0.4 V on a Rh-film electrode in a N2-saturated 0.1 mol/L KOH solution(B)[43], optimized structure of FeN3-graphene(C)[45],high-angle annular dark-field scanning transmission electron microscopy(HAADF-STEM)image of Mo0/GDY sample(D)[50](B) The reference spectrum was taken at 0.4 V. (A,B) Copyright 2020,Wiley-VCH; (C) Copyright 2016,American Chemical Society; (D) Copyright 2019,American Chemical Society.
1.2.2 非贵金属催化剂 由于贵金属催化剂用于NRR电催化合成NH3的效果仍不理想,一些过渡金属同样具有能接受N2孤电子对的空d轨道,同时因价格低廉、 能够通过“π反馈”的反应机制活化N≡N键而被广泛关注. Fe是自然界固氮酶中成分最多的元素之一,也是传统Haber-Bosch工艺的常用催化剂,研究人员推测Fe在电催化NRR中具有良好的催化活性. Li等[45]结合第一性原理设计了石墨烯负载的不同过渡金属单原子催化剂,高自旋极化的FeN3中心[图3(C)]相比其它过渡金属(钪、 钒、 锰等)催化剂显示出更强的吸附与活化N2能力,能够有效提高电催化NRR合成氨的效率. Hu等[46]对比了不同价态Fe用于电催化NRR合成NH3的活性,发现具有混合价态的铁/四氧化三铁(Fe/Fe3O4)催化剂对于电催化活性有显著影响,并且选用PBS能够有效抑制HER活性,在-0.3 V(vs. RHE)下产生NH3的法拉第效率可达8.29%(约为Fe箔的120倍). Chu等[47]使用Fe掺杂二氧化铈(CeO2)时,发现CeO2从晶体状纳米颗粒转变为含有大量VO的非晶态纳米片时暴露出更多的活性位点,加速了电子传输. DFT计算进一步表明Fe 与相邻的VO共存时,Ce3+-Ce3+对是促进NRR 反应和抑制HER 反应的重要活性位点,显著提高了NRR合成NH3的性能,NH3产率为26.2 mg·h-1·mg-1,法拉第效率为14.7%. Mo元素同样是固氮酶的重要活性组分之一,Mo基催化剂通常比Fe基催化剂具有更好的电催化NRR活性. 通过对不同晶面的纳米Mo 晶体进行DFT 计算[48],分析得知在Mo(110)面,N 原子比H 原子吸附能力更强,更利于合成NH3,NRR活性更强; 而在Mo(211)面,HER副反应占主导地位,NRR合成NH3的活性降低,并通过控制电化学沉积时间制备具有不同Mo(110)面含量的Mo催化剂,得知Mo(110)面暴露最多时催化剂用于电催化NRR的起始电位最低,且法拉第效率最高. 因此,控制催化剂的晶面形貌能够改变几何结构和电子结构,是提高纯金属NRR 合成NH3活性的重要途径. 研究人员还常使用暴露活性位点、 控制形貌、 掺杂元素和调控电子结构来提高Mo 基催化剂的NRR 合成NH3的活性[49~51]. Zeng 等[49]开发了富含硫(S)空位的N 掺杂的MoS2纳米花,掺杂的N 元素促进了S 空位的产生,形成的Mo—N 键加速了N2活化,提高了NRR 电催化合成NH3的法拉第效率(9.1%). Hui 等[50]制备了具有NRR 和HER 活性的石墨二乙炔上锚定零价单原子Mo催化剂(Mo0/GDY)的双功能电催化剂[图3(D)],在中性电解质中均具有良好的催化活性(145.4 µg·h-1·mg-1)和法拉第效率(21.0%). Zhong 等[51]在MoO2纳米颗粒上构建了丰富的氧空位,氧空位诱导电子转移使其作为高效NRR吸附与活化位点,进一步提高了NH3合成的法拉第效率(27.6%). 钨(W)金属因弱H 原子结合能力能够有效减弱HER 而增强NRR,具有良好的合成NH3潜力. Wang等[52]制备了WO3与C3N4异质结(WO3-C3N4-R)催化剂,向WO3引入氧空位能够增大费米能级,从而诱导更多电子由WO3转移至C3N4形成增强的界面电场,通过强W—N 键结合使N≡N 极化断裂,加速NRR动力学过程.
1.2.3 非金属催化剂 尽管金属基NRR电催化剂占主导地位,但因存在低活性位点利用率、 较低负载量和易被氧化还原的缺点仍限制了其在NRR电催化合成NH3反应中的应用. 非金属催化剂因通常具有较大的比表面积、 丰富的价电子与空位缺陷和较弱吸附H 原子能力,能够提供更为理想的N2活化中心,提高反应活性和选择性,引起研究人员的关注[23,53~56]. 最近,碳基材料被认为是NRR合成NH3的新型有效电催化剂. 如Liu等[54]通过热解沸石咪唑酯骨架(ZIF-8)制备N掺杂多孔碳催化剂(NPC),改变制备温度以调节催化剂中不同N物种的含量,通过对比得知,在750 ℃下制备得到的NPC-750含有的吡啶N、 吡咯N 和石墨N 分别占比6.2%,5.3%和2.1%,相比于分别在850 和950 ℃下制备得到NPC-850和NPC-950具有更高的催化合成NH3活性(1.4 mmol·g-1·h-1)和选择性. DFT计算表明,其良好的性能是由于材料中含有较多的吡啶N和吡咯N吸附与活化位点用于合成NH3,并且分析其合成NH3路径为*N≡N→*NH=NH→*NH2-NH2→2NH3. 同时,NPC 的大表面积(432.0 cm2/g)和三维连通的网络结构在提高电催化NRR合成NH3催化活性方面起着重要作用. 前者有利于N2的吸附,后者为分子扩散和电子传输提供了运输通道. 研究表明,通过构建多孔隙率结构和调整杂原子含量能够显著提高电催化NRR活性. 硼(B)元素的缺电子性和低电负性使其在电催化NRR合成NH3反应中具有良好的催化活性和选择性. Yu等[55]开发了B掺杂石墨烯催化剂(BG)并用于电催化NRR合成氨,局域缺电子性的掺杂B 能够高效吸附弱路易斯碱N2分子,BC3结构有效降低了氨合成的反应能垒,具有较高的合成氨产率(9.8 µg·h-1·cm-2)和法拉第效率(10.8%). Zhang 等[56]制备了二维的B 纳米片材料(BNS),通过DFT 计算解释了BNS 对于提高电催化NRR 活性的作用机制(图4),结果表明: (1) N2在3B(自旋极化磁矩为0 µB)处得到有力吸附,通过缔合机制的远端配位形式合成NH3,N≡N 键由1.112 Å(1 Å=0.1 nm)拉长到1.132 Å[图4(A)]; (2) 差分电荷分析表明,3B 向N2分子转移了0.38 个电子用于活化N2分子[图4(B)]; (3) B(104)表面氢化后成为电催化NRR 还原合成NH3的有效活性位点,抑制HER 的发生[图4(C)]; (4) B的2p轨道与*N2的2p轨道有部分重叠,B原子的sp3杂化能够增强B—N反馈键,从而增加N—N 的π*反键轨道[图4(D)和(E)]. 表1 总结了不同催化剂用于电催化NRR 合成NH3的性能数据.

Table 1 Summary of performance of electrocatalytic NH3 synthesis by NRR
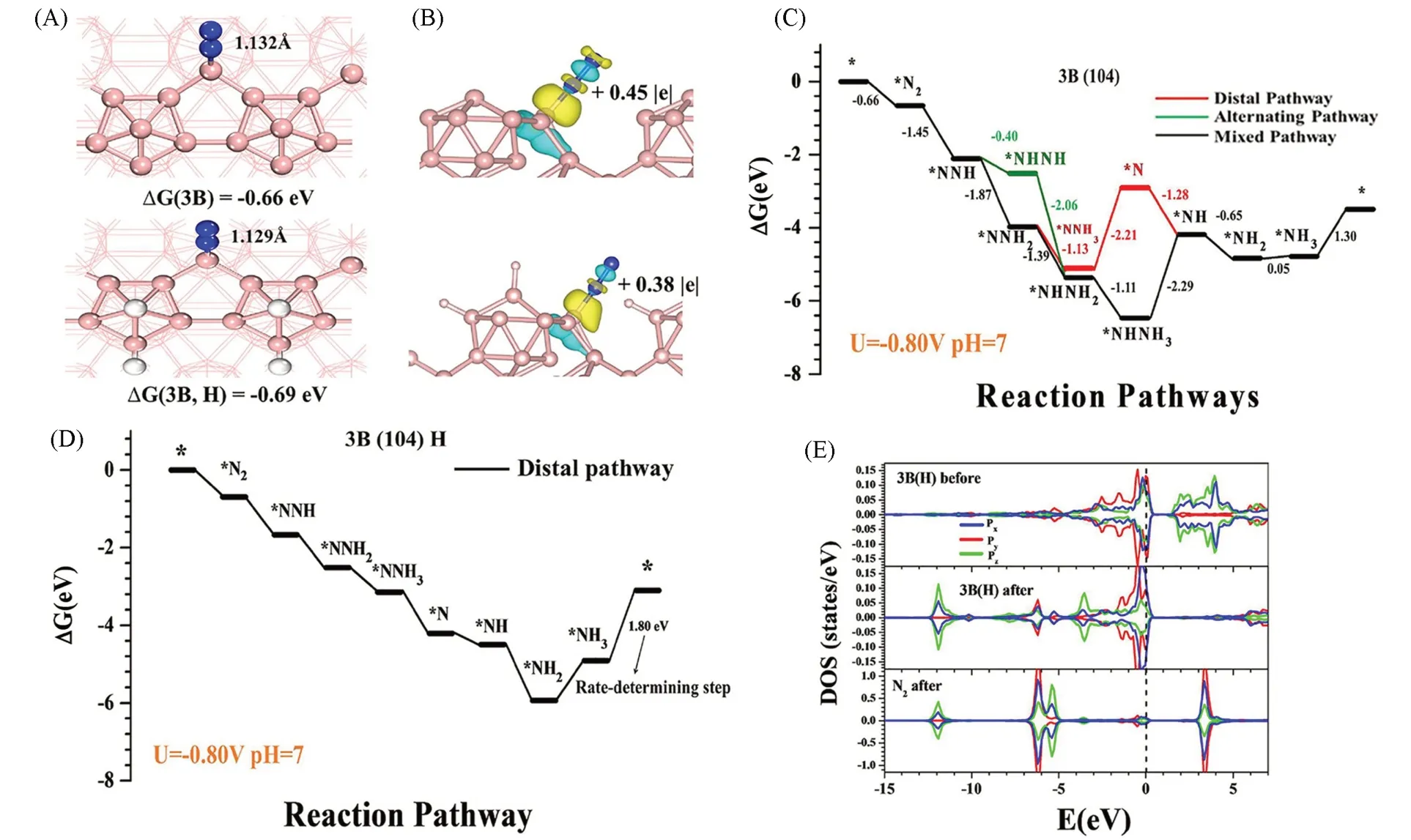
Fig.4 Optimal structure of N2 adsorption with end-on coordination on 3B and 3B(H) sites(A), the electron density difference after N2 adsorption on 3B and 3B(H) sites(B), the corresponding free-energy diagram of NRR on the B(104) surface at U=-0.80 V(C, D), the density of state(DOS) of 3B(H) site before and after N2 adsorption(E)[56](A,B) Yellow and blue colors represent electron accumulation and depletion,respectively,and the isosurface level is 0.01 e/Bohr; (E) legend: B: pink; N: blue; H: white. Copyright 2019,American Chemical Society.
1.3 电催化NRR合成NH3小结
近年来,在水溶液电解质中的NRR合成NH3由于绿色环保、 成本低、 设备简单等优势引起了研究人员的广泛关注,但仍存在产生NH3速率低和法拉第效率低的问题. 本文围绕目前电催化NRR 合成NH3的大量进展,将催化剂总结为贵金属催化剂、 非贵金属催化剂和非金属催化剂3类,能够有效归纳总结出电催化NRR合成NH3反应体系和催化剂设计的机理与思路. 贵金属催化剂表面极强的结合能和优异电子传输能力在电催化NRR 反应中得到了大量研究,但因价格昂贵和效果仍不理想限制了其应用,若通过调控金属存在与分散的形式和选择合适的载体将贵金属减量化并提高电催化NRR合成NH3性能,有望得到进一步应用. 研究人员在对自然界和Haber-Bosch工艺固氮基础上进行探究,一些价格低廉的非贵金属催化剂同样具有能够接受N2孤电子对的空d轨道并用于电催化NRR合成NH3,通过提高活性位点暴露率、 控制形貌、 掺杂元素和调控电子结构可有效提高电催化NRR合成NH3活性和选择性. 非金属基催化剂也得到了较为深入的探索,能够有效解决金属催化剂中活性位点利用率低、 金属负载量低和易被氧化还原的问题. 在反应机理探索方面,各个原位表征手段能够原位动态检测反应中的物种演变和材料结构变化,通过多种技术手段相互佐证能够明晰电催化NRR合成NH3的动力学和机理的深层次数据,为开发高效高选择性NRR电催化剂提供有效指导. 目前,理论计算多用于与实验工作结合在微观尺度上研究电催化NRR合成氨的反应路径、 活性中间体能垒、 催化剂表面电子转移等,对于电催化NRR合成NH3体系中底物质量传输速率、 溶解度和温度等因素不够重视,需进一步精进电催化NRR合成NH3的计算模型与方法,为设计高效NRR电催化剂的精细结构提供理论指导.
2.1 电催化还原反应合成NH3机理

Fig.5 Mechanism of electrocatalytic NO3RR for NH3 synthesis
2.2 电催化NO3RR合成NH3性能调控
选用合适高效的催化剂在调控电催化NO3RR合成NH3性能方面起着关键作用. 催化剂活性通常受到活性位点处的微观电子结构和暴露数量的影响,调控活性位点的电子结构可以通过缺陷工程、 界面工程、 合金效应、 异质原子掺杂等策略完成,控制活性位点暴露数量可以通过构建单原子催化剂或构建特殊形貌与结构的催化剂等实现. 此外,还可以通过以上催化剂设计策略调控电催化合成氨有利步骤的进行和抑制HER副反应的进行,使反应具有更高的产率和选择性.
2.2.1 缺陷工程 构筑催化剂表面的空位缺陷能够调节催化剂的电子结构,为提供吸附和活化位点,提高催化效率[65,66]. Jia等[67]构建富含Vo的TiO2纳米管用于高法拉第效率(85.0%)和高选择性合成NH3(87.1%),通过DEMS 和DFT 计算确定了硝酸盐的氧原子填补到TiO2-x的Vo处削弱N—O 键来抑制副产物的产生. Wei等[68]对平均粒径为25 nm的锐钛矿晶型和金红石晶型的TiO2(P25 TiO2)进行煅烧构建氧缺陷后作为阴极[图6(A)],发现在600 ℃下H2还原得到的P25 TiO2-600用于电催化NO3RR的转化率(86.3%)、 NH3选择性(81.9%)和法拉第效率(78.0%)明显高于P25 TiO2(转化率为55.5%、NH3选择性为60.6%、 法拉第效率为51.0%),结合电子顺磁共振(EPR)和傅里叶变换红外光谱(FTIR)表征发现,对能够显著提高反应活性,VO为提供了吸附位点促进还原转化. DFT计算表明,在TiO2异相结上构建氧缺陷可以有效降低的吸附能,促进*NOH中间体的形成,最终获得更高的NH3合成效率. Du等[69]使用静电纺丝法合成具有均匀分布丰富Vo的钛酸铁(Fe2TiO5)纳米纤维,Vo的加入提高d带中心至更高的水平,Fe 原子与氧(O)原子结合形成低于费米能的反键态,加速催化剂与中间体之间的电荷转移,在0.1 mol/L 硝酸钠(NaNO3)的PBS 电解液中,法拉第效率达到87.6%,NH3产率达到1.36 mmol·h-1·mg-1. Huang等[70]报道了聚合物石墨相氮化碳(g-C3N4)催化剂,通过调控催化剂N 空位数量以优化电催化合成NH3性能,达到了较高的法拉第效率(89.9%)和选择性(69.8%).15N 同位素标记实验表明,NH3的合成完全来源于,排除了空气中N 源和催化剂本身含N 的污染[图6(B)]. DFT计算表明,适量的N空位能够在催化剂的费米能级引入新电子态,并调节NO3RR中间体的吸附能(*NO,*NOH,*NH2等),促进的吸附与活化,提高NH3合成的效率. 因此,在催化剂载体或自身引入O,N等缺陷,能够有效调节NO3RR合成NH3的性能.
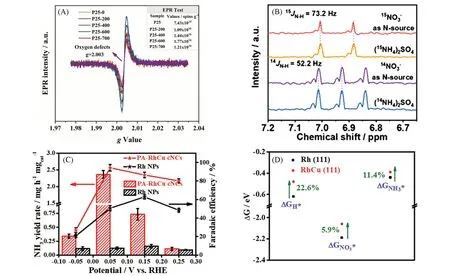
Fig.6 EPR spectra of P25-M(M=0, 200, 400 and 600)(A)[68], 1H NMR spectra of the electrolyte using and as the reactant(B)[70], NH3 yield and Faradaic efficiency of NH3 for -RR at PA-RhCu cNCs and Rh NPs at different potentials(C) and ΔGH*, ΔGNO3*, and ΔGNH3* on Rh(111) and RhCu(111) surfaces(D)[83](A) Inset table is referred to corresponding spin quantum numbers. Copyright 2022,Elsevier; (B) Copyright 2021,American Chemical Society; (C,D) Copyright 2022,Wiley-VCH.
2.2.2 界面工程 设计构筑具有强界面效应的NO3RR电催化剂是提高合成NH3性能的重要途径[71]. 由两个或多个组分组成的电催化剂组分间的强界面相互作用对电催化过程起着至关重要的作用,不同组分间的界面结构理论上可以建立起组分间的电子或反应中间体的高速传输通道,并诱导电子耦合效应,有助于提高电催化过程中催化剂的活性和稳定性[72,73]. Yin 等[74]在氧化亚铜(Cu2O)纳米片上沉积银纳米颗粒(Ag/Cu2O)作为阴极,达到高电催化NO3RR催化活性(选择性96.9%、 法拉第效率96.5%).在线DEMS和DFT计算表明,Ag纳米颗粒作为吸附位点与Cu2O之间的强界面相互作用促进电荷转移,有效抑制*NOH生成的同时促进*N的生成,以提高NH3合成的选择性. Yu等[75]开发了具有异质结构的钴/氧化钴(Co/CoO)纳米片催化剂,界面间存在的肖特基势垒促使电子由Co转移至CoO导致Co的缺电子态,提高了副产物生成的能量势垒,抑制了HER竞争反应,表现出优异的NO3RR合成NH3的法拉第效率(93.8%)和选择性(91.2%). Sun等[76]将氯化亚铜(CuCl)(111)和金红石型TiO2(110)层叠,通过分子动力学模拟和有限元分析表明,TiO2向CuCl 发生电子转移形成的内置电场模型引起了催化剂周围的聚集,加速了的传质过程,最终NH3产率为1.82 mg·h-1·cm-2.
2.2.3 合金效应 向金属催化剂中引入其它元素构建合金效应能够通过各元素间的协同作用和电子结构调制作用调控中间产物与反应路径,为改善催化活性提供了一种合理的策略[77~79]. Fang等[80]模仿具有双功能性质的铜型亚硝酸盐还原酶开发了CuCo 合金纳米片催化剂,其中,Cu具有较强的吸附能力,Co作为电子-质子转移的活性位点还原,CuCo双金属具有优异的调控*H和*NO3中间体作用,在-0.2 V(vs. RHE)下NH3合成的法拉第效率高达(100±1)%. Wang等[81]引入Fe金属改善Ru金属表面易被活性氢(*H)覆盖导致难以吸附与转移电子至的π*反键轨道的问题,制备了超薄二维Ru-Fe合金纳米结构加快了反应动力学,发现Ru 4d轨道与Fe 3d轨道的互补加速电子转移与维持价态稳定,能够达到38.68 mg·h-1·的NH3产率和20 次循环的优异稳定性. Yin 等[82]利用介孔空心球结构PdCu/Cu2O 催化剂中的PdCu 合金效应促进NO3RR 反应,Pd 的引入使得Cu 3d轨道发生极化,促进电子转移(Pd 3d→Cu 3d),*Pd 活性位点为吸附提供了空轨道,*Cu 加速了还原,NH3选择性达96.7%,法拉第效率为94.3%. 在线DEMS 和DFT 计算的相互验证表明,PdCu 合金抑制*NOH 中间体形成的同时促进*N 中间体的的形成,提高了催化性能. Gao 等[83]将RuCu 合金负载于氧化还原石墨烯(RuxCuy/rGO),合金效应调节了金属的d带中心位置及和的吸附能,Cu 位点高效还原为,Ru位点则高效还原为NH3,在-0.05 V(vs. RHE)的超低电位下,NH3合成速率为0.38 mmol·cm-2·h-1、法拉第效率为98%[图6(C)和(D)]. Liu等[84]提出构建具有介孔结构的金属-非金属合金的PdX纳米立方催化剂,具有96.1%的NH3合成法拉第效率和3760.0 µg·h-1·mg-1的产率. N的合金化调节了Pd表面电子结构,形成大量的低配位活性位点,有利于吸附和NH3解吸,降低了反应所需总能量,从动力学上加速了NO3RR电催化合成NH3反应,同时显著抑制了Pd的扩散和浸出,增强了催化剂的稳定性.
2.2.4 单原子催化剂 具有近100%原子利用率的单原子催化剂中独特配位环境的活性位点均匀分布,具有优异的催化活性和选择性,已成为电催化反应中理想的催化剂[85,86]. Wu等[87]将Fe以单原子形式分散于N掺杂碳基底(Fe SACs),分散的金属位点能够避免副产物N2合成过程中的N—N耦合步骤,提高了NH3合成的法拉第效率(75%). DFT 计算表明,*NO 是电催化NO3RR 的关键中间体,通过对比Fe SACs,Co SACs 和Ni SACs 催化剂上的还原极限电位[0.3 V(vs. RHE),0.42V(vs. RHE)和0.39 V(vs. RHE)],可知Fe SACs 的低热力学势垒大大提高了NH3合成的产率(20 mg·h-1·mg-1).Wang等[88]构建了一种N配位的Cu-Ni双单原子催化剂(Cu/Ni-NC)[图7(A)和(B)],该催化剂与Cu单原子催化剂(Cu-NC)和Ni 单原子催化剂(Ni-NC)相比具有更高的催化活性和选择性. 在线DEMS 和原位衰减全内反射表面增强红外吸收光谱检测到反应过程中的关键中间体(*NO2,*NO,*NH)[图7(C)],推断出NO3RR 合成NH3的反应路径. DFT 计算表明,Cu-Ni 双单原子间的轨道杂化和电子转移(Ni→Cu)有利于的吸附与活化,并且有效降低限速步骤的能量势垒、 抑制N—N耦合,提高了NH3合成产率与选择性. 该课题组还使用氢溢流的方法制备了Au/Cu 单原子合金负载于Cu2O(Au/Cu SAAs)[89],在-0.80 V(vs. RHE)电位下表现出高NH3合成的选择性(96.48%)和法拉第效率(99.69%). 采用傅里叶变换对Au L3边k3加权的扩展X射线吸收精细结构(Extended X-ray absorption fine structure,EXAFS)谱进行分析,由2.4 Å处的显著峰确定了Au/Cu SAAs中存在Au-Cu配位. DFT计算表明,Au单原子能有效地降低吸附能,Au—Cu键的存在促进了*NO2向*NO的还原过程,提高了NO3RR合成NH3的选择性. Zhang 等[90]通过电替换和脱合金过程构建了具有Cu 空位的Au1Cu(111)单原子合金(VCu-Au1Cu SAAs),Au单原子与相邻的Cu空位协同调控了催化剂表面的局域电子结构和几何结构,加速了H2O活化产生*H,在动力学上加速结合*H的还原反应与产物*NH3的解吸,显著提高了合成NH3的法拉第效率(98.7%).
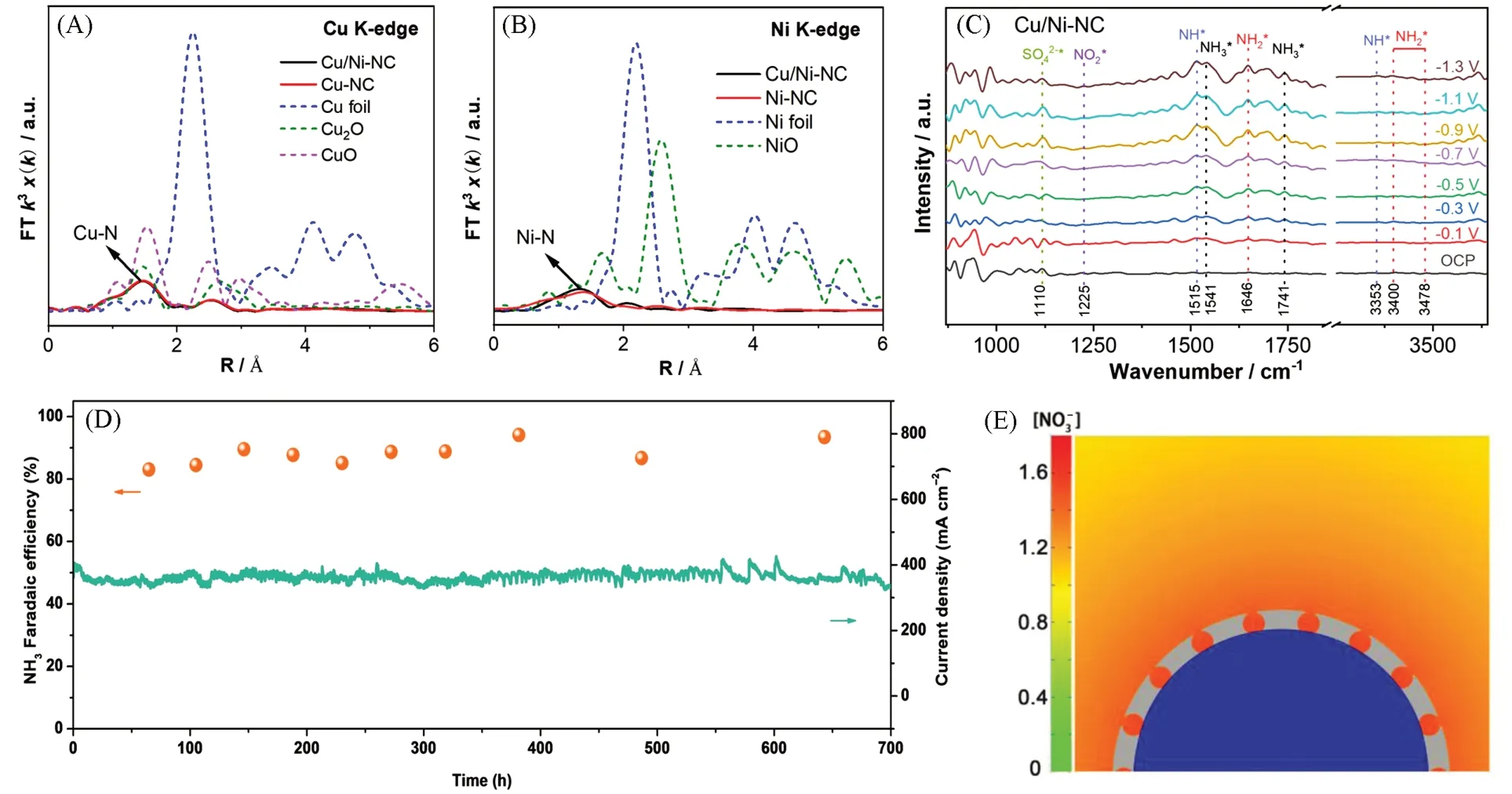
Fig.7 FT magnitude of the experimental EXAFS spectra of Cu/Ni-NC and references at Cu K-edge(A) and Ni K-edge(B), respectively, in situ ATR-SEIRAS spectra of Cu/Ni-NC from OCP to -1.3 V(vs.RHE) in electrolytes containing 0.1 mol/L Na2SO4 and 100 mg/L NO3-N(C)[88], time-dependent NH3 Faradaic efficiency and current density over Cu nanosheets during long-term stability test in a course of 700 h(D)[96], simulated concentrations and distributions of local on the surface of Cu encapsulated with porous carbon at the diffusion time of 7 µs(E)[97]Copyright 2023,Wiley-VCH.
2.2.5 异质原子掺杂 异质原子掺杂能够调制催化剂的电子结构和改变几何结构,进而调控对反应中间体的吸附强度和反应路径,提高电催化反应活性. 通常将其分为金属元素掺杂和非金属元素掺杂.Wu等[91]使用密度泛函理论对含N物种(*NO3和*NO2)在不同金属掺杂Cu催化剂上各步骤反应的能垒进行计算,发现Zn 掺杂能够调节*NO 吸附构型转变为桥式吸附、 提高中间体的吸附强度并降低反应能垒,得到的NH3合成法拉第效率和产率分别为98.4%和5.8 mol·g-1·h-1. Zhang 等[92]使用Co 掺杂的Fe-MOF-74作为前驱体制备了Co掺杂的Fe@Fe2O3催化剂(Co-Fe@Fe2O3),Co取代部分Fe位点调节催化剂的d带中心以调整中间产物的吸附能并抑制HER 副反应,产生NH3的选择性达到(99.0±0.1)%.Zhu等[93]开发Ru/β-Co(OH)2催化剂减弱Ru活性位点与中间体*NH3物种的d-p轨道杂化,使其能够有效脱附,有效促进了NO3RR反应动力学,NH3产率达0.38 mmol·cm-1·h-1. 非金属元素(N,B,P等)掺杂碳材料也已被广泛研究,掺杂原子与碳原子间的电负性差异能够改变催化剂的电子结构和缺陷位点,使其具有良好的电催化性能. Li等[94]使用模板法构建多孔聚苯胺水凝胶,通过改变煅烧温度调控得到的碳气凝胶催化剂中N 掺杂的种类和含量,N 掺杂由于会在碳骨架上造成电荷分布不对称,对电催化NO3RR合成NH3具有显著影响. 结合DFT计算分析了对NO-3吸附能和反应自由能,可知石墨N基团有利于吸附和降低*NO到*HNO步骤的质子化能量势垒,具有1.3 mg·h-1·cm-2的NH3产率和95.0%的法拉第效率.
2.2.6 形貌结构调控 可控的构建具有特定形貌与结构的催化剂同样有利于定向调控反应路径,提高氨合成的效率和选择性. Huang等[95]使用激光照射聚酰亚胺聚合物,根据动力学效应合成了含有应变和无序的五边形、 六边形和七边形的非晶态石墨烯(ox-LIG),应用于流动型电解池(Flow cell)中在-0.93 V(vs. RHE)电位下NH3合成,得到的法拉第效率和产率分别高达约100%和2859.0 µg·cm-2·h-1.通过X射线对分布函数分析和电子显微镜表征明确了催化剂的非晶石墨烯的独特形貌和成键方式. 原位FTIR光谱和DFT计算明确了石墨烯非晶态结构在结构弛豫作用稳定反应中间体形成NH3的关键作用. Fu等[96]开发了由CuO纳米片原位制备具有(100)面和(111)面的Cu纳米片催化剂,将其应用于流动型电解池中具有优异的氨合成性能和700 h 运行的高稳定性[图7(D)]. 电化学测试和DFT 计算表明,Cu不同晶面的静电势差异导致了中间体吸附强度差异,Cu(100)面易吸附与活化生成,随后转移至Cu(111)面进一步还原合成NH3. Song 等[97]将Cu 纳米颗粒封装在多孔碳框架中(Cu@C)并用于高效电催化还原超低浓度的合成NH3,采用有限元法模拟了在Cu@C和Cu表面的富集程度[图7(E)],发现多孔碳的富集作用使得Cu@C催化剂表面明显积聚,-0.3 V(vs. RHE)电位下实现了高效(产率为469.5 µg·h-1·cm-2)和高法拉第效率(72.0%,为Cu催化剂的3.6倍)电催化NO3RR合成NH3. 表2总结了不同催化剂用于电催化NO3RR合成NH3的性能数据.
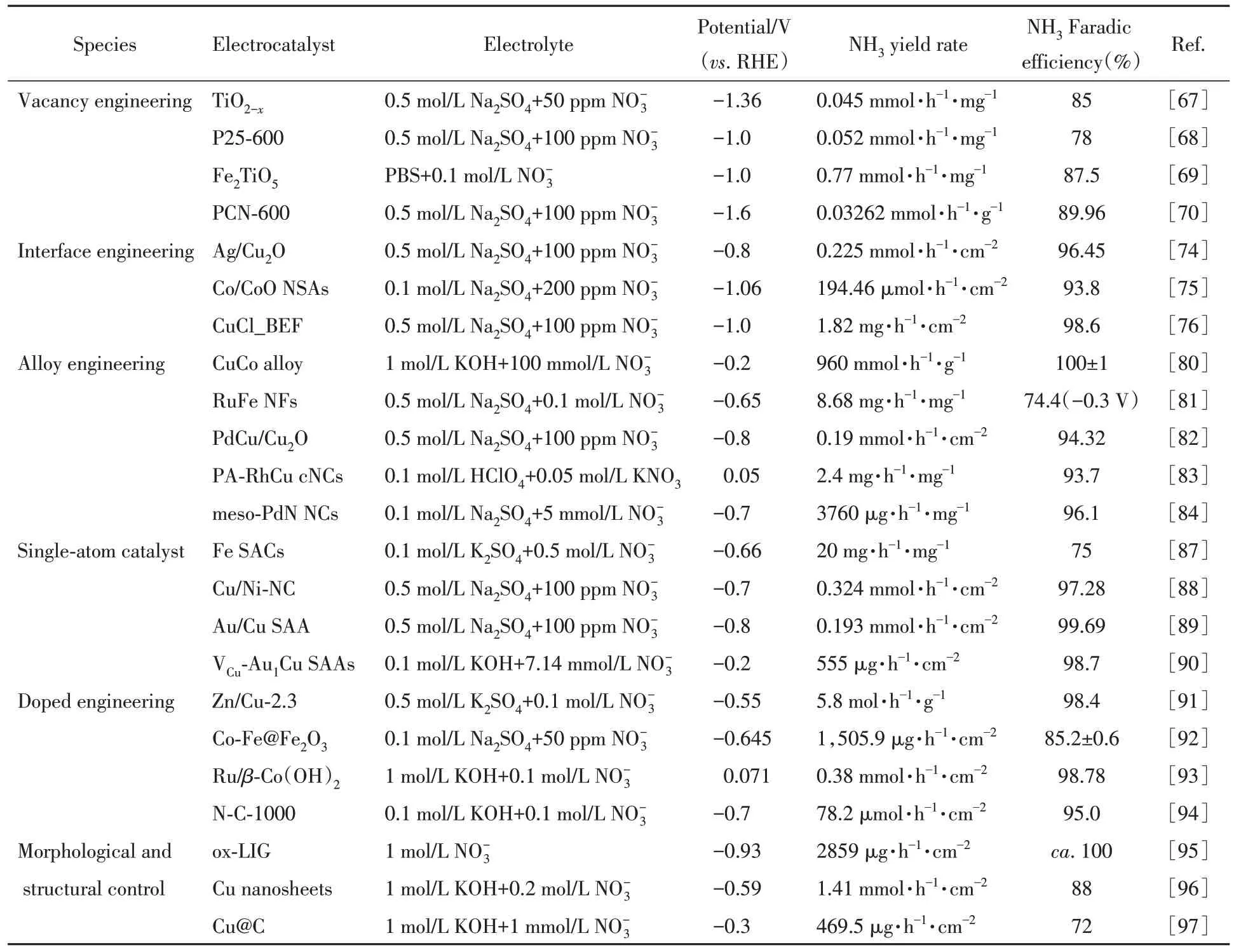
Table 2 Summary of performance of electrocatalytic NH3 synthesis by NO3RR
2.3 电催化NO3RR合成NH3小结
与电催化NRR相比,电催化NO3RR因能够克服气体传质影响、 反应能垒低和N=O键更易解离等优点,有望实现更高效高选择性合成NH3,并且能够同时有效解决环境污染和能源短缺的问题. 若设计催化剂精细结构调控活性位点处电子结构和暴露数量能够有效克服反应路径复杂和析氢副反应导致其目前活性与选择性较低的问题. 本章综述了缺陷工程、 界面工程、 合金效应、 异质原子掺杂、 单原子催化剂和构建催化剂特殊形貌和结构的催化剂调控策略在电催化NO3RR合成NH3方面的应用. 构建催化剂表面空位缺陷能够有效调节催化剂的电子结构、 调控d带中心位置、 促进电子转移至,以增强对的吸附与活化; 通过界面工程构筑电催化剂的多组分间电子和反应中间体的快速传输通道,并诱导电子耦合效应,有助于提高催化剂活性和稳定性; 构建合金效应能通过各元素间协同效应和电子结构调制作用调节中间产物与反应路径; 异质原子掺杂能够调制电子结构和几何结构,以调控对反应中间体的吸附强度和反应路径,提高反应活性; 单原子催化剂的超高原子利用率和独特化学环境的活性位点能够有效增强催化活性和选择性; 可控地构建特性形貌和结构的催化剂也有利于定向调控反应路径,增强电催化NO3RR合成氨的活性、 选择性、 法拉第效率和稳定性. 在催化剂制备过程或活性评价过程中借助先进的原位操作表征手段检测催化剂与活性物种的动态变化,能够全面了解不同精细结构催化剂上进行的复杂反应步骤,有利于加深对反应动力学机理和催化剂合成的认识,进一步推动电催化NO3RR合成NH3领域的发展. 理论计算已被视为良好的催化剂筛选工具,但仍需注意反应中的关键参数(pH、 电解液种类与浓度、 温度等)的选取,并与实验结果结合,为设计出更高效、 高选择性、 高稳定性的NO3RR合成氨催化剂起到良好的指导作用.
3 以NO为N源
3.1 电催化NO还原反应合成NH3机理
NO是最常见的空气污染物和温室气体之一,主要来源于汽车尾气、 化石燃料与工厂废气[98,99]. NO不仅会造成酸雨、 导致水体富营养化,同时也会与人体血液中血红蛋白结合危害人类生命健康[100~102].目前,选择性催化还原(SCR)技术以NH3或H2为还原剂,将NO转化为无害的N2是最主要的NO去除方式,但该工艺存在操作成本高、 产物附加值低、 会造成NH3逃逸等缺点[103~105]. 与电催化NRR相比,电催化NO还原反应(Nitrogen oxide reduction reaction,NORR)因极性分子NO键能较弱更易发生反应,因此选用高效高稳定性的电催化剂能够有效提高NH3的合成效率. Long 等[106]基于中间体吸附能的标度关系和描述符的方法,选用N吸附自由能GAD(N)作为NORR描述符,通过DFT计算筛选出Cu作为高选择性产生NH3的电催化NORR 催化剂. 并确定NORR 反应机理分为解离和缔合两种机制(图8)[106]:解离机制中NO分子键首先断裂后分别氢化为NH3和H2O; 缔合机制中NO首先被氢化为中间体HxNOHy后被连续还原为NH3和H2O,共分为远端氧加氢、 远端氮加氢、 交替氮加氢和交替氧加氢4种途径. 为了验证理论计算,他们选用铂(Pt)箔、 Cu箔和泡沫铜作为电催化剂转化NO合成NH3,选用泡沫铜作为电极时,催化剂在-0.9 V(vs. RHE)条件下产生NH3速率达到517.1 µmol·h-1·cm-2,法拉第效率为93.5%. 相比之下,Pt箔的产NH3效率和法拉第效率也较低. 同时计算动力学势垒表明,合成NH3路径比合成N2O,N2和H2路径在热力学上更为可行.

Fig.8 Associative mechanism of electrocatalytic NORR for NH3 synthesis
3.2 电催化NORR合成NH3性能调控
Ko 等[107]研究了Fe,Co,Ni,Cu,Pd,Ag 和Pt 等一系列过渡金属催化剂[图9(A)和(B)],在气体扩散电极(GDE)流动型电解池中进行NORR,使用0.1 mol/L 氢氧化钠(NaOH)和0.9 mol/L 氯酸钠(NaClO4)作为电解液,发现电催化NORR 的产物选择性相近,说明这些催化剂上的反应机理相似,其中,Cu对电催化NORR合成NH3选择性最高,与Long等[106]的研究结果一致. 并且该课题组深入研究了NO分压和pH值对Cu催化剂用于NORR的影响,发现在催化剂表面NO覆盖率低时,NH3选择性更高,反之,则易发生N-N耦合生成N2与N2O; 在酸性电解质中,NH3合成的选择性明显提高,法拉第效率接近100%. Chen 等[108]提出将Cu 以金属单原子的形式分散于MoS2载体并用于电催化NORR,在-0.6 V(vs. RHE)电位下表现出最高的NH3产率(337.5 µmol·h-1·cm-2)和法拉第效率(90.6%),远高于电催化NRR的效率; 而当电位低于-0.6 V(vs. RHE)时,HER竞争反应增强,NORR选择性降低. 通过DFT计算可知,在反应过程中,0.4 个e 由Cu 单原子转移到*NO 的未填充轨道上,用于活化和裂解N=O 键.同时,通过在线DEMS明确了*NH2OH中间体(m/z=33)的存在,推断出该催化剂采用混合加氢的路径合成NH3(*NO→*NHO→*NH2O→*NH2OH→*NH2→*NH3). Dhanabal 等[109]进一步将Fe,Co,Ni,Cu,Zn 等过渡金属纳米颗粒包埋于一维碳纳米棒(TM-NCNR)探究石墨化、 孔隙率和掺杂N原子的影响,在发现在700 ℃煅烧下Ni-NCNM催化剂中Ni位点与掺杂N原子的协同作用表现出良好的电催化NORR活性.Zhao等[110]设计合成了5种不同晶面曲线的单晶Ni箔,并研究了其对电催化NORR活性影响,得知高指数面(210)能够降低关键中间体氢化决速步骤的能量势垒,在流动型电解池中表现出较高的NH3产率(544.0 µmol·h-1·cm-2)和法拉第效率(>85%),稳定运行时间超过50 h. 尽管d区过渡金属元素含有未充满的价层d轨道可以有效吸附和活化NO分子,但也有利于结合质子促进竞争性的HER,不利于提高电催化NORR 选择性; 与之相比,主族p区金属的弱质子结合能力能够有效减弱HER 副反应的竞争.Chen等[111]将p区金属锑(Sb)单原子负载于非晶态MoO3基底,Sb的掺入能够有效调整MoO3电子结构,减小带隙宽度,加速NORR电催化合成NH3中的电子传递和反应动力学,在-0.6 V(vs. RHE)时表现出较高NH3产率(273.5 µmol·h-1·cm-2)和法拉第效率(91.7%). 理论计算表明,Sb 位点能够增强对*NO/*NHO的吸附,降低电催化NORR的反应能垒,减弱对H2O的吸附能力.

Fig.9 Total current density vs. potential of various catalysts(A), Faradaic efficiency of various catalysts at(0.10±0.02) V(vs. RHE) in NO electroreduction(B)[107], diagram of the flow electrolyzer for NORR over Ru-LCN(C)[115], reaction process of NORR on the hcp-Co with the reaction free energy and the optimized structures of all reaction intermediates(D), partial density of states(PDOS) of d orbitals in the hcp-Co and fcc-Co(E)[118](E) The Fermi energy level is set to 0 eV. (A,B) Copyright 2022,American Chemical Society; (C) Copyright 2022,American Chemical Society; (D,E) Copyright 2023,American Chemical Society.
Langer 等[112,113]将Pt,Rh,Ru 等贵金属催化剂用于流动型电解池研究酸性溶液中的电化学NORR合成NH3过程,发现了电催化NORR还原产物很大程度上依赖于电极电位,当电极表面没有吸附氢时,主要产物为N2O,但当电极电位更负时,还原产物会由N2,NH2OH转向生成NH3. 并且发现在低电位下控制NO流速能够改变NO与中间体在电极表面的吸附,当NO流速较慢时,NO与催化剂表面更长时间地接触并产生更多的还原物种,有利于提高NH3产率. 但由于NO 分子的腐蚀性,金属电催化剂在与NO长时间接触过程中常会被腐蚀溶解,从而限制电催化剂在电催化NORR过程中的长期使用稳定性.为了解决这一问题,Muthusamy 等[114]制备了在碳纤维上(CF)包裹的镍纳米颗粒的N 掺杂碳催化剂(NiNC@CF),独特的核壳状CF与Ni纳米颗粒活性相的强烈电子相互作用阻碍了Ni纳米颗粒的腐蚀溶解,在-0.5 V(vs. RHE)电位下表现出更佳的NH3选择性(法拉第效率为87.0%).
为了解决实际应用中NO低浓度(<5%)的问题,Li等[115]对高配位数的Ru纳米片进行等离子体处理制备了具有强NO吸附能力的低配位数Ru纳米片,在-0.2 V(vs. RHE)电位下,该催化剂具有较高的法拉第效率(65.9%)和产率(45.1 µmol·h-1·mg-1),远高于Cu 催化剂(法拉第效率为8.2%). 通过DFT 计算对比了Ru(ΔGNO=-2.35 eV)和Cu(ΔGNO=-1.0 eV)上的吸附吉布斯自由能,证明Ru具有优异的NO富集能力,在低浓度NO条件下能够提高NORR性能. 同时,该课题组选用自制GDE[图9(C)]以适应工业中连续生产中测试该催化剂电催化NORR性能(NH3产率为84.2 µmol·h-1·mg-1),远高于在H型电解槽中的性能. Cheon 等[116]向电极炭黑中混合纳米级零价铁在1% NO 气氛还原中达到高NH3合成产率(1239.0 µmol·cm-2·h-1)和法拉第效率(96.0%).
研究人员在提高NORR合成NH3活性方面同样运用了诸多的催化剂结构调控策略. Zhang等[117]将石墨片与硫化钼(MoS2)纳米片结合(MoS2/GF),发现该催化剂中带正电荷Mo空位增强对NO的吸附与活化,并且能够抑制结合质子以抑制HER 反应,提升电催化NORR 合成NH3的产率与选择性. Wang等[118]制备了具有高电催化NORR活性和稳定性的六方Co纳米片(hcp-Co),运用DFT计算和NO程序升温脱附实验表明,优异活性来源于催化剂独特的电子结构和质子穿梭效应[图9(D)和(E)],NH3产率和法拉第效率分别达439.5 µmol·h-1·mg-1和72.6%. Peng等[119]制备了B/N共掺杂于碳纳米管的铌(Nb)单原子催化剂,Nb金属的d轨道与NO的π*轨道能级发生杂化有利于NO分子在Nb位点的吸附,随后电子由Nb位点转移至NO 使N—O键延长至1.2 Å促进加氢过程,NH3产率高达8.2 × 10-8mol·cm-2·s-1(接近美国能源部目标: 9.3×10-7mol·cm-2·s-1). Li 等[120]设计了将富含氧缺陷TiO2负载于Ti 板(TiO2-x/TP)催化剂,相比TiO2更利于NO 脱氧与质子化过程,在0.2 mol/L PBS 电解液中表现高NH3产率(1233.3 µg·h-1·cm-2). Liang 等[121]将具有高导电性和高耐腐蚀性的非晶态氮化硼(a-B2.6C)沉积于负载在Ti板的TiO2(a-B2.6C@TiO2/Ti)催化剂,a-B2.6C层的B—C键易向NO的π2p*轨道转移电子活化NO,有效提高NH3合成产率(3678.6 µg·h-1·cm-1)和法拉第效率(87.6%). Shao 等[122]利用理论计算通过全局热力学优化研究了6种具有不同晶体结构的Cu-Sn合金,选择了合金Cu6Sn5制备气体扩散电极,在电流密度>1400 mA/cm2时,NH3合成产率可达10 mmol·cm-2·h-1,法拉第效率可达96%; 在包含膜电极组件的电解槽中电流为400 A,电压为2.6 V(vs. RHE)时,NH3合成产率可达到2.5 mol/h,具有良好的工业化前景. 表3总结了不同催化剂用于电催化NO3RR合成NH3的性能数据.

Table 3 Summary of performance of electrocatalytic NH3 synthesis by NORR
3.3 电催化NORR合成NH3小结
与NRR相比,极性分子NO的键能更弱更易被活化,设计合适的反应体系和催化剂精细结构有助于高效高选择性合成NH3. NO是常见的气体污染物,目前的NO去除方法成本高、 产物附加值低、 会造成NH3逃逸,若能通过电催化NORR方法高效高稳定性合成NH3,既有助于解决废气污染问题,也能带来巨大的能源和经济价值. 目前,在电催化高浓度NORR合成NH3方面,研究人员已针对NO分压、 NO流速、 体系pH 值、 反应器设计、 催化剂结构调控等多种因素进行了较为深入的探索,有效提高了NORR 合成NH3的产率和选择性. 但实际应用中废气体系复杂,NO 浓度低,若使用富集装置富集NO会大大增加成本,阻碍其商业化应用,将研究重点转向低浓度NO 或实际工业废气中的NORR 势在必行. 同样,运用先进的动态微观表征手段明晰反应机理和活性位点演变,并通过详尽全面的理论计算手段筛选催化剂结构机理调控能够有效指导电催化NORR合成NH3体系的设计与建立,使该领域得到进一步的发展.
4 总结与展望
近几年来,电催化NRR 合成NH3的路线已成为研究热点,与传统NRR 合成NH3的Haber-Bosch 工艺相比,电催化NRR合成NH3能在小型设备中实现,从而支持低能耗分布式NH3与化肥的生产. 但目前该过程中仍存在N2在水中溶解度低、 难以控制析氢反应等问题,其产NH3速率及法拉第效率仍难以满足工业化生产. 解决这些问题应从以下几个方面出发: 首先,应调控活性位点处的电子结构、 增加活性位点数量以优化低成本催化剂活性,同时优化电极本身结构,改进负载催化剂电极附近的质子与电子浓度,控制反应步骤,抑制析氢副反应,提高合成NH3的效率和选择性. 其次,应解决N2扩散传质限制的问题,通过优化反应器结构和电解液的组成为工艺的运行提供稳定的工作环境,提高反应中N2溶解程度以增强传质过程,并调控电解液中质子和电子的传输性能抑制副反应.
与NRR相比,电催化NO3RR能够有效处理环境水体中存在的污染,也能够广泛应用于污水处理,为有效解决环境水体污染实现资源化利用提供了一条重要的途径. 相比于N2,作为N源易溶于水,能够大大减轻传质的影响,且N=O 键更易解离,能够减少能量消耗,更易被活化. 但电催化NO3RR合成NH3过程更为复杂,反应中间体种类众多,条件更为苛刻. 若能够选择合适的催化剂调控策略设计催化剂的精细结构,通过缺陷工程、 界面工程、 合金效应、 界面效应、 单原子分散、 异质原子掺杂和构建特定形貌与结构的催化剂等手段调控材料表面结构、 电子结构和活性位点数量,调整反应步骤削弱副反应过程,能够达到反应在较低还原电位下稳定高效高选择性合成NH3的目的.
与NRR相比,电催化NORR能够有效解决汽车尾气与工厂尾气中的NO污染排放问题,为有效解决环境大气污染实现资源化利用提供了一条重要的途径. 电催化NORR 中极性分子NO键能较弱,更易被吸附与活化,选用合适的反应体系与构建催化剂精细结构有望实现高效高稳定性NH3合成. 但实际应用中车辆、 发电厂和化工厂排放的NO废气浓度低、 常含有SOx与有机化合物等多种杂质,不同氮氧化物间也存在转换,NO易被氧化生成二氧化氮,反应存在诸多影响因素. 若能在后续研究中探究诸多影响因素的干扰、 完善反应体系、 调整反应器和电解质种类,有望降低合成NH3所需的电势,提高电流效率,促使反应向高效高选择性合成NH3的方向进行.
在反应机理探索方面,运用各个原位表征手段能够清晰跟踪检测到反应过程中的活性物种演变和材料结构变化,各种手段相互佐证能够得到反应动力学和机理的深层次数据,后续研究中需开发更多的原位技术,为设计高效NRR电催化剂提供有效指导. 通过N同位素标记实验进行对比,能够排除反应底物其它N源的干扰,进一步明确反应的各个步骤与机理. 目前,理论计算能从分子或原子水平计算反应各步骤的自由能与能量势垒的变化、 催化剂表面电子结构等,但仍需全面考虑底物质量传输速率、 溶解度、 pH值和温度等因素,在探索反应步骤与机理的同时精准筛选反应体系条件与催化剂精细结构,为高效合成NH3提供理论指导与思路.
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:48
应用化工(2020年9期)2020-09-29 08:55:16
小学生必读(高年级版)(2019年3期)2019-06-21 06:50:28
中国有色金属学报(2018年2期)2018-03-26 07:58:37
天天爱科学(2017年2期)2017-04-29 00:44:03
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中国资源综合利用(2016年7期)2016-02-03 03:00:13
电源技术(2015年9期)2015-06-05 09:36:06
故事会(2015年12期)2015-05-14 15:24:30
中南民族大学学报(自然科学版)(2014年4期)2014-08-06 05:49:24
