
新型4-双取代吡唑啉酮合成子的合成
2024-03-23 00:56:58蒋静,李阳,黄维,李想
合成化学 2024年3期
蒋 静,李 阳,黄 维,李 想
(成都中医药大学 药学院,西南特色中药资源国家重点实验室,四川 成都 611137)
吡唑啉酮类化合物是一类重要的五元氮杂环化合物[1],在诸多药物分子或功能材料中普遍存在[2-4],广泛应用于药物化学、有机合成以及农业化学等方面[5-6]。在化学领域,吡唑啉酮类化合物可作为一种功能强大的合成子,具有多取代、多活性位点的特性[7]。以C4-位不同取代的吡唑啉酮为底物,构建结构多样的吡唑啉酮衍生物,对于新型药物的研发和功能材料的开发具有重要意义[8-11]。
目前,C4-位不同取代的吡唑啉酮合成子主要可分为4-无取代吡唑啉酮,4-单取代吡唑啉酮和具有α,β-不饱和键的吡唑啉酮,其中具有α,β-不饱和键的吡唑啉酮又可分为α,β-不饱和吡唑啉酮、插烯吡唑啉酮、吡唑啉二酮和吡唑啉酮亚胺。这些合成子可应用于Michael加成、Friedel-Crafts反应和Conia-ene环化反应等各类型反应,进而构建出非环取代、螺环取代或并环稠合的吡唑/吡唑啉酮衍生物(图1a)[12-15]。然而,关于4-双取代吡唑啉酮合成子的研究相对较少,还有待进一步开发。

图1 C4-位不同取代的吡唑啉酮合成子及其衍生物
为了进一步丰富吡唑啉酮合成子的种类,更大程度地增加化合物的多样性,本研究尝试构建了新型的4-双取代吡唑啉酮合成子(图1b)。基于4-无取代的吡唑啉酮α-碳原子上的活泼氢,首先在碱作用下使之形成烯醇化合物,再于N-氯代丁二酰亚胺(NCS)作用下,高效合成4-氯取代的4-双取代吡唑啉酮。
1 实验部分
1.1 仪器与试剂
BUCHI M-565型熔点仪;Bruker-600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent G1969-85000型质谱仪。
所用试剂均为分析纯。
1.2 合成
将化合物1(5.00 mmol)和氢氧化钙(0.74 g,10.00 mmol)加入到圆底烧瓶中,以1,4-二氧六环(10 mL)为溶剂,在80 ℃中反应40 min,再加入对硝基苯基氯甲酸酯(1.06 g,5.10 mmol),继续反应30 min,通过TLC监测反应完毕后,将烧瓶移至室温冷却,加入2M HCl将pH调至3~4,加入水(20 mL)和二氯甲烷(20 mL),萃取分离各相,水相用二氯甲烷(2×20 mL)萃取,合并有机相,用饱和食盐水(20 mL)洗涤,无水硫酸钠干燥,过滤,将滤液真空浓缩可得化合物2。
将化合物2(4.00 mmol)、0.4 nm分子筛(0.50 g)、甲苯(8 mL)和羟胺化合物(4.00 mmol)依次加入圆底烧瓶中,在80 ℃下反应,通过TLC监测反应进程。待反应完毕后,将反应液真空浓缩,经硅胶柱层析(洗脱剂:二氯甲烷∶甲醇=1∶1,V∶V)纯化得化合物3。
将化合物3(2.90 mmol)加入到圆底烧瓶中,以二氯甲烷(5 mL)为溶剂,冰浴下加入N-氯代丁二酰亚胺(384.00 mg,2.90 mmol),通过TLC监测反应进程。约5 min即可反应完毕,将反应液真空浓缩,经硅胶柱层析(洗脱剂:二氯甲烷∶甲醇=5∶1,V∶V)纯化得化合物4。
将化合物N-(苄氧基)-4-氯-3-甲基-5-氧代-1-苯基-4,5-二氢-1H-吡唑-4-甲酰胺(4a)(52.70 mg,0.15 mmol)和1,3-二甲基吲哚(5)(14.50 mg,0.10 mmol)加入到反应试管中,再加入碳酸钾(27.60 mg,0.20 mmol),以三氟乙醇(1 mL)为溶剂,常温下反应,通过TLC监测反应进程。约3 h即可反应完毕,将反应液真空浓缩,经硅胶柱层析(洗脱剂:石油醚∶乙酸乙酯=10∶1,V∶V)纯化得化合物6(21.60 mg),收率46%。
4a:白色固体,收率88%; m.p.134.0~133.5 ℃;1H NMR(600 MHz,CDCl3)δ:9.31(s,1H),7.75(d,J=7.2 Hz,2H),7.37~7.30(m,7H),7.17(d,J=7.2 Hz,1H),4.87(q,J=10.8 Hz,2H),2.27(s,3H);13C NMR(151 MHz,CDCl3)δ:165.2,157.5,155.5,135.8,133.0,128.6,128.2,128.0,127.7,125.2,118.1,77.8,60.5,13.2; HR-MS(ESI-TOF)m/z:Calcd for C18H17ClN3O3{[M+H]+}358.0958,found 358.0959。
N-(苄氧基)-4-氯-1,3-二甲基-5-氧代-4,5-二氢-1H-吡唑-4-甲酰胺(4b):白色固体,收率91%; m.p.120.6~122.7 ℃;1H NMR(600 MHz,CDCl3)δ:9.30(s,1H),7.50~7.22(m,3H),4.89(q,J=10.8 Hz,2H),3.24(s,3H),2.16(s,3H);13C NMR(151 MHz,CDCl3)δ:168.2,158.6,155.9,134.1,129.6,129.2,128.7,78.8,59.9,32.0,14.1; HR-MS(ESI-TOF)m/z:Calcd for C13H15ClN3O3{[M+H]+}296.0802,found 296.0801。
N-(苄氧基)-4-氯-3-甲基-1-(萘-2-基)-5-氧代-4,5-二氢-1H-吡唑-4-甲酰胺(4c):白色固体,收率66%; m.p.159.1~161.3 ℃;1H NMR(600 MHz,CDCl3)δ:9.32(s,1H),8.22(s,1H),7.91(dd,J=9.0 Hz,1.8 Hz,1H),7.86~7.73(m,3H),7.42(dt,J=10.8 Hz,7.2 Hz,2H),7.34(dd,J=25.2 Hz,5.4 Hz,5H),4.89(q,J=10.8 Hz,2H),2.32(s,3H);13C NMR(151 MHz,CDCl3)δ:166.4,158.5,156.7,134.4,134.0,133.3,131.5,129.7,129.3,129.0,128.8,128.1,127.7,126.8,126.0,118.1,116.9,78.9,61.6,14.4; HR-MS(ESI-TOF)m/z:Calcd for C22H19ClN3O3{[M+H]+}408.1115,found 408.1116。
N-(苄氧基)-4-氯-1-(3-氟苯基)-3-甲基-5-氧代-4,5-二氢-1H-吡唑-4-甲酰胺(4d):白色固体,收率76%; m.p.140.1~143.0 ℃;1H NMR(600 MHz,CDCl3)δ:9.27(s,1H),7.57(dd,J=18.0 Hz,8.4 Hz,2H),7.36~7.26(m,6H),6.87(td,J=8.4 Hz,3.0 Hz,1H),4.87(q,J=9.6 Hz,2H),2.26(s,3H);13C NMR(151 MHz,CDCl3)δ:166.2,163.6,162.0,158.3,156.8,138.2,138.2,134.0,130.4,130.3,129.7,129.3,128.8,114.3,114.3,113.0,112.8,106.6,106.4,78.9,61.8,14.2; HR-MS(ESI-TOF)m/z:Calcd for C18H16ClFN3O3{[M+H]+}376.0864,found 376.0862。
N-(苄氧基)-4-氯-1-(3-氯苯基)-3-甲基-5-氧代-4,5-二氢-1H-吡唑-4-甲酰胺(4e):白色固体,收率71%; m.p.137.4~141.0 ℃;1H NMR(600 MHz,CDCl3)δ:9.22(s,1H),7.82(s,1H),7.70(dd,J=7.8 Hz,1.8 Hz,1H),7.36~7.31(m,5H),7.27(t,J=7.8 Hz,1H),7.15(dd,J=7.8 Hz,1.8 Hz,1H),4.87(q,J=10.8 Hz,2H),2.26(s,3H);13C NMR(151 MHz,CDCl3)δ:166.2,158.2,156.8,137.9,134.8,133.9,130.1,129.7,129.3,128.8,126.2,119.1,116.8,78.9,61.7,14.2; HR-MS(ESI-TOF)m/z:Calcd for C18H16Cl2N3O3{[M+H]+}392.0569,found 392.0566。
N-(苄氧基)-4-氯-3-甲基-5-氧代-1-(对甲苯基)-4,5-二氢-1H-吡唑-4-甲酰胺(4f):白色固体,收率79%; m.p.133.0~135.7 ℃;1H NMR(600 MHz,CDCl3)δ:9.32(s,1H),7.61(d,J=8.4 Hz,2H),7.36~7.30(m,5H),7.14(d,J=8.4 Hz,2H),4.87(q,J=10.8 Hz,2H),2.28(s,3H),2.26(s,3H);13C NMR(151 MHz,CDCl3)δ:165.1,157.5,155.5,135.1,133.4,133.0,128.6,128.5,128.2,127.7,118.2,77.8,60.3,20.0,13.3; HR-MS(ESI-TOF)m/z:Calcd for C19H19ClN3O3{[M+H]+}372.1115,found 372.1112。
N-(苄氧基)-4-氯-3-乙基-5-氧代-1-苯基-4,5-二氢-1H-吡唑-4-甲酰胺(4g):白色固体,收率85%,m.p.151.0~154.1 ℃;1H NMR(600 MHz,CDCl3)δ:9.29(s,1H),7.78(d,J=8.4 Hz,2H),7.33(dd,J=21.6 Hz,6.6 Hz,7H),7.16(d,J=7.2 Hz,1H),4.87(q,J=11.4 Hz,2H),2.63(dddd,J=25.2 Hz,18.0 Hz,10.8 Hz,7.2 Hz,2H),1.26(t,J=7.8 Hz,3H);13C NMR(151 MHz,CDCl3)δ:166.4,160.3,158.6,137.0,134.0,129.7,129.3,129.0,128.8,126.2,119.1,78.9,62.0,21.8,9.2; HR-MS(ESI-TOF)m/z:Calcd for C19H19ClN3O3{[M+H]+}372.1115,found 372.1119。
N-(苄氧基)-4-氯-3-环丙基-5-氧代-1-苯基-4,5-二氢-1H-吡唑-4-甲酰胺(4h):白色固体,收率69%,m.p.138.5~141.3 ℃;1H NMR(600 MHz,CDCl3)δ:9.31(s,1H),7.73(d,J=7.8 Hz,2H),7.43~7.24(m,7H),7.15(t,J=7.2 Hz,1H),4.89(q,J=12.6 Hz,2H),1.86(s,1H),1.19~1.15(m,2H),1.19~0.10(m,3H);13C NMR(151 MHz,CDCl3)δ:166.1,161.4,158.6,137.0,134.1,129.7,129.2,129.0,128.8,126.1,119.1,78.8,62.1,9.9,9.4,8.8; HR-MS(ESI-TOF)m/z:Calcd for C20H19ClN3O3{[M+H]+}384.1115,found 384.1110。
N-(苄氧基)-4-氯-1-(4-氯苯基)-3-甲基-5-氧代-4,5-二氢-1H-吡唑-4-甲酰胺(4i):白色固体,收率73%; m.p.152.5~153.9 ℃;1H NMR(600 MHz,CDCl3)δ:9.33(s,1H),7.75(d,J=8.4 Hz,2H),7.35(t,J=7.2 Hz,2H),7.29(s,4H),7.18(d,J=14.4 Hz,1H),4.84(q,J=10.8 Hz,2H),2.28(s,3H);13C NMR(151 MHz,CDCl3)δ:165.2,157.6,155.5,135.8,134.2,131.5,129.9,128.0,127.9,125.3,118.1,76.9,60.4,13.3; HR-MS(ESI-TOF)m/z:Calcd for C18H16Cl2N3O3{[M+H]+}392.0569,found 392.0562。
1′-(苄氧基)-3,3a′,8′-三甲基-1-苯基-1′,3a′,5′,8a′-四氢-2′-H-吡唑-4,3′-吡咯并[2,3-b]吲哚]-2′,5(1H)-二酮(6):白色固体,收率46%; m.p.266.1~272.7 ℃;1H NMR(600 MHz,CDCl3)δ:7.54(d,J=6.0 Hz,2H),7.46~7.40(m,2H),7.38~7.29(m,3H),7.25(t,J=6.0 Hz,2H),7.15~7.04(m,2H),6.71(d,J=6.0 Hz,1H),6.58(t,J=12.0 Hz,1H),6.46(d,J=6.0 Hz,1H),5.23(d,J=12.0 Hz,1H),5.01(d,J=12.0 Hz,1H),4.63(s,1H),2.98(s,3H),2.23(s,3H),1.41(s,3H);13C NMR(151 MHz,CDCl3)δ:167.1,161.1,154.9,149.3,136.3,133.4,128.8,128.8,128.6,128.2,127.7,127.6,124.1,122.3,118.0,117.7,106.6,87.3,77.1,66.8,51.2,33.9,24.1,16.9; HR-MS(ESI-TOF)m/z:Calcd for C28H27N4O3{[M+H]+}467.2083,found 467.2088。
2 结果与讨论
2.1 反应条件优化
以化合物1a参与的反应进程为模板,对反应条件进行了优化。首先对化合物2的合成过程进行优化(表1)。以1,4-二氧六环为溶剂,在60 ℃下对碱进行筛选。结果发现:尽管在加入三乙胺、氢氧化钾和氢氧化钙时都能以良好的收率得到目标化合物,但当使用氢氧化钙作为碱时,收率相对较高,因此将其作为优势碱参与下一步对反应溶剂的筛选。结果显示:将1,4-二氧六环更换为其他溶剂如二氯甲烷和四氢呋喃时,收率明显降低。在此基础上继续对反应温度进行筛选,从表1中可以看出,收率随着温度的升高明显升高。由此确定化合物2的最优合成条件为:以氢氧化钙为碱,1,4-二氧六环为溶剂,在80 ℃下反应。

表1 化合物2合成条件的优化
接着优化了化合物3和4的合成过程,分别依次对反应中涉及到的溶剂和温度进行筛选(表2~3)。结果发现,在化合物3的合成过程中,甲苯为最优溶剂,升高温度能够促进反应进行,80 ℃下收率较好。在化合物4的合成过程中,不同溶剂条件下均不能有效提高收率。接着对反应温度进行考察,发现温度能显著影响反应进程,将温度降至0 ℃时,能使收率最大化。因此确定化合物3的最优合成条件为:以甲苯为溶剂,在80 ℃下反应;化合物4的最优合成条件为:以二氯甲烷为溶剂,在0 ℃下反应。
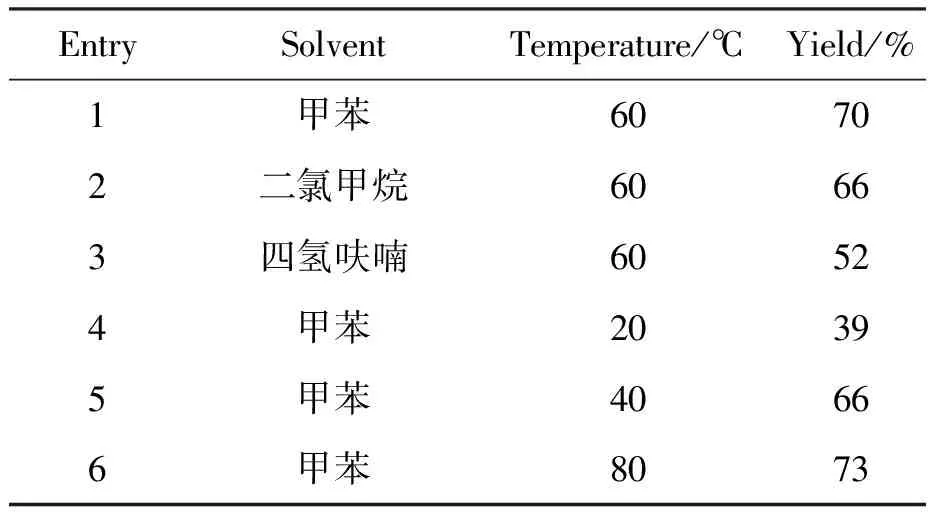
表2 化合物3合成条件的优化

表3 化合物4合成条件的优化
2.2 普适性考察
在筛选优化后的条件下,对反应底物的普适性进行了考察(图2)。分别以不同取代的吡唑啉酮和羟胺化合物为底物进行反应,均能以良好到优秀的收率(66%~91%)分离得到相应的目标化合物(4a~4i),证明该合成策略具有良好的底物普适性。
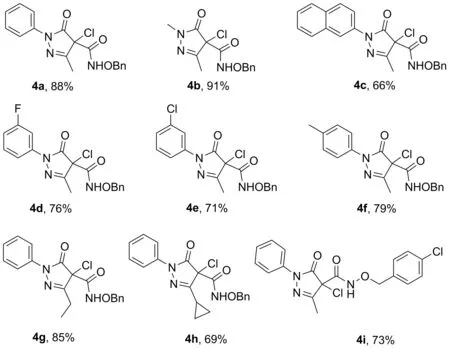
图2 底物的普适性考察
2.3 合成子的应用
为了进一步验证该合成子的应用价值,继续探究了其参与环化反应的可能性。在碳酸钾作用下,化合物4a通过与1,3-二甲基吲哚(5)之间的1,3-偶极反应,顺利构建出新型[3+2]环加成产物6(图3)。该反应过程条件温和、反应速度快且收率(46%)与立体选择性良好(3∶2dr),体现了新型4-双取代吡唑啉酮合成子在化学合成方面的应用潜力。

图3 4-双取代吡唑啉酮合成子在环化反应中的应用
3 结论
本研究以4-无取代吡唑啉酮为前体,顺利合成了新型的4-双取代吡唑啉酮合成子。通过对反应条件的筛选优化和对底物普适性的考察,以温和的反应条件和良好的产物收率(66%~91%)合成出9个不同取代的4-氯取代的4-双取代吡唑啉酮化合物。同时,该化合物作为合成子进一步参与1,3-偶极反应,能够以46%的收率顺利构建出新型[3+2]环加成产物,成功丰富了吡唑啉酮合成子的种类,增加了化合物的多样性。
猜你喜欢
兰州文理学院学报(自然科学版)(2024年2期)2024-06-12 00:00:00
化学与粘合(2023年6期)2023-12-15 17:16:06
今日农业(2021年2期)2021-11-27 19:19:53
今日农业(2020年23期)2020-12-31 09:00:40
山东化工(2019年11期)2019-06-26 03:26:44
现代农业(2016年4期)2016-02-28 18:42:14
合成化学(2015年1期)2016-01-17 08:59:30
中南民族大学学报(自然科学版)(2015年2期)2015-12-16 12:11:10
应用化工(2014年1期)2014-08-16 13:34:08
化工生产与技术(2014年3期)2014-02-27 13:41:44
