
甲基2,3-O-磺酰-α-D-葡萄糖苷合成5-羟甲基糠醛(5-HMF)及其衍生物的新方法
2024-03-23 00:56张菡旭李敏娇李端华陈国华
合成化学 2024年3期
张菡旭,李敏娇,杨 蓉,李端华,陈国华*
(1.四川轻化工大学 化学工程学院,四川 自贡 64300; 2.成都大学 药学院,四川 成都 610106)
随着传统石油资源的日渐枯竭,如何从生物质出发,再通过化学转化方式获得替代燃料资源和发展新型化工原料品已成为当今生物质资源化利用研究的重点[1-2]。纤维素是自然界中分布最广、含量最多的一种多糖,占植物界碳含量的50%以上,其在强酸条件下水解,可以90%以上收率获得D-葡萄糖单体。纤维素廉价易得,且用其制备D-葡萄糖单体方法有着简单,污染少,收率高等优点。这些优点使其成为一种重要的可再生性生物质来源。因此,如何利用廉价易得的D-葡萄糖单体为起始原料制备一些具有重要应用价值的基础化工品有着重要的研究意义[3]。现今,基于D-葡萄糖单体为起始原料的生物质转化研究主要集中在转化为5-羟甲基糠醛(5-HMF)的平台化合物上,因为该化合物可以制备一些广泛应用于医药、塑料和燃料等领域的高附加值的化工品中[4-8],如2,5-呋喃二甲酸、2,5-二甲酰呋喃[2]、2,5-二甲基呋喃、乙酰丙酸和1,6-己二醇等。其中,2,5-呋喃二甲酸是合成聚呋喃二甲酸乙二醇酯(PEF)的主要单体[9-10],其比现年产量超过7000万吨的聚对苯二甲酸乙二醇酯(PET)的性能更加优越,但是由于5-HMF存在价格高昂、制备过程中副产物多和难以达到聚合单体纯度要求的缺点,限制了PEF的进一步发展[11-12]。
如何高效地从D-葡萄糖单体中制备5-HMF一直是生物质转化研究的重点,到目前为止该方面的研究一直都是当前研究的热点[3-10]。综合分析发现:现有的制备研究主要集中在2条基础路线上,第一条是先将纤维素先效水解制备出D-葡萄糖单体,然后再采用“一锅法”制备5-HMF;第二条是直接利用果糖为起始原料,一锅法制备。但无论是利用D-葡萄糖还是果糖为起始原料制备,其研究的重点都集中于在特定的反应溶剂体系中,直接高温脱水制备不同类型的催化剂。该制备过程的关键转化在于单糖结构的2-位羟基的羰基化,从而获得关键中间体3-脱氧葡萄糖醛酮(图1),然后经过由吡喃糖环到呋喃糖环的缩环化,获得5-HMF的呋喃环化结构,最终对应化合物经过高温脱水化形成目标物[13]。虽然一些文献报道的制备产率并不低[3,5-6,10],但这些方法一旦应用到连续型的工业化生产中时,由于糖类的多羟基结构特点,会使得每一单元反应都可能发生许多副反应,生成大量的不可预知的副产物,与此同时还会生成大量的水。这些不利因素会导致精心制作的各类型催化剂迅速失效,无法在工业中实现连续型的工艺生产,进一步会影响到催化剂的再生。同时,5-HMF的熔点低,且沸点亦高,在分离纯化时,由于生成的结构类似的副产物与5-HMF相溶性好,采用现有常规分离纯化方法,获得高纯产品更是不易[6]。这些问题都是当前工业化直接利用D-葡萄糖单体制备5-HMF时面临的困境,因此导致5-HMF,特别是高纯的5-HMF价格高昂,限制了其作为基础平台化合物进一步的工业化应用。因此打破传统研究思路,研究新的5-HMF合成方法是一个具有应用前景的课题,具有重要的经济价值。基于前期的研究发现:一个易由D-葡萄糖单体制备的衍生物——甲基2,3-O-磺酰-α-D-吡喃葡萄糖苷(图2),能够高效地制备出5-HMF,这为以D-葡萄糖为底物合成5-HMF提供了很好的方法学支持。
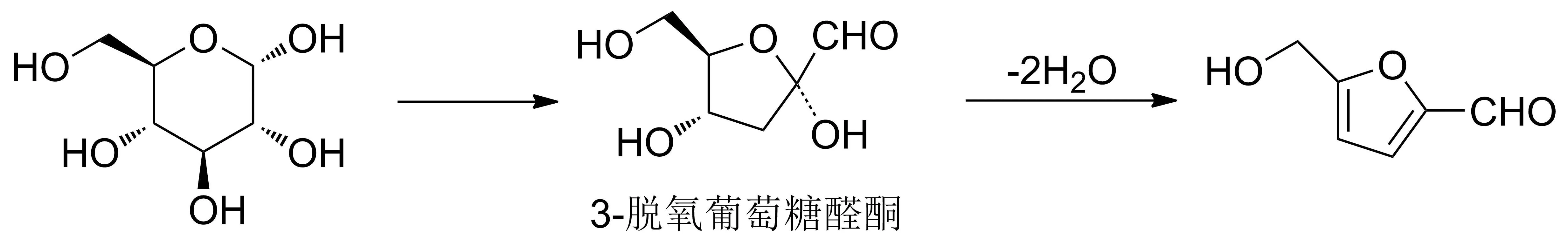
图1 D-葡萄糖催化脱水制备5-HMF的基本反应途径
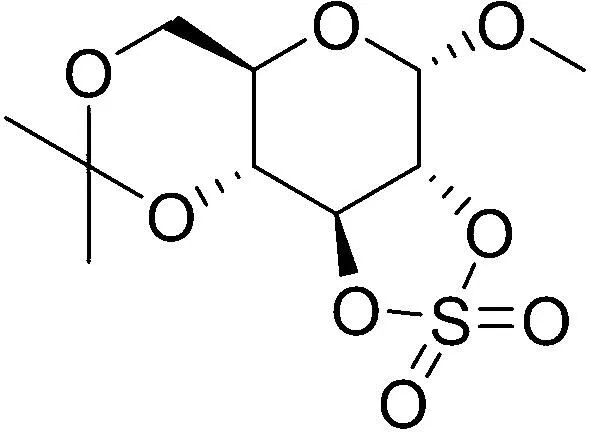
图2 甲基2,3-O-磺酰-α-D-吡喃葡萄糖苷的结构
1 实验部分
1.1 仪器与试剂
BRUKER ASCEND 600型核磁共振波谱仪(CDCl3为溶剂,TMS为内标),柱层析硅胶(200-300目,青岛美高集团有限公司);GF254型硅胶板(德国Merck公司)。
甲基-α-D-葡萄糖苷(≥98%,阿拉丁试剂有限公司);磺酰氯(≥98%,上海泰坦科技股份有限公司);苯甲酰氯(≥98%,上海泰坦科技股份有限公司);其他均为国产分析纯试剂。
1.2 合成
(1) 甲基4,6-亚丙基-2,3-O-环磺酸酯基-α-吡喃葡萄糖苷(化合物Ⅰ)的合成
参考文献[14]方法制备,产率75%;1H NMR(600 MHz,CDCl3)δ:5.12(d,J=3.0 Hz,1H),5.08(t,J=9.6 Hz,1H),4.59(dd,J=10.2 Hz,3.0 Hz,1H),4.12(t,J=9.6 Hz,1H),3.93~3.85(m,2H),3.76~3.72(m,1H),3.52(s,3H),1.54(s,3H),1.44(s,3H);13C NMR(150 MHz,CDCl3)δ:100.32,96.01,80.82,80.72,71.98,66.17,61.83,56.13,28.72,19.02。
(2) 2,3-O-环磺酸酯基-甲基-α-D-吡喃葡萄糖苷(化合物Ⅱ)的合成
按化学计量比称量化合物Ⅰ(15.0 g,0.0506 mol)加入圆底烧瓶(150 mL)中,随后加入甲醇(75 mL)的溶剂中,再滴加5滴质量分数为98%的浓硫酸,常温(15~25 ℃)下搅拌反应3 h,TLC监测反应完全后(展开剂:乙酸乙酯),再加入5%的NaHCO3水溶液调节pH至中性,旋转蒸干回收甲醇,剩余物中加入300 mL乙酸乙酯溶解,用100 mL饱和食盐水洗涤分液,有机相经无水硫酸钠干燥,过滤,减压蒸馏得到白色固体约12.2 g,产率94.0%;1H NMR(600 MHz,DMSO-d6)δ:5.75(s,2H,OH),5.24(d,J=1.8 Hz,1H),4.93~4.97(m,2H),3.95~3.99(m,1H),3.69(dd,J=12.0 Hz,1.2 Hz,1H),3.59(dd,J=12.0 Hz,5.4 Hz,1H),3.44~3.46(m,1H),3.41(s,3H);13C NMR(150 MHz,CDCl3)δ:94.61,86.19,81.43,75.24,67.22,59.89,55.41。
(3) 甲基2,3-O-环磺酰基-6-O-乙酰基-α-D-吡喃葡萄糖苷(化合物Ⅲ)的合成
按化学计量比称量化合物Ⅱ(10.0 g,0.0390 mol)加入圆底烧瓶(250 mL)中,随后加入冰乙酸60 mL,充分溶解后,滴入5滴浓硫酸,常温搅拌反应6 h,TLC监测反应完全后(展开剂:石油醚∶乙酸乙酯=7∶3,V∶V),向反应完全的混合体系中加入150 mL乙酸乙酯稀释,该有机相用水(2×100 mL)洗涤分液,合并水相。水相再用50 mL乙酸乙酯反萃取,合并有机相,有机层用100 mL饱和食盐水洗涤,分液,得到有机相,经无水硫酸钠干燥,过滤,减压蒸馏得到白色粗产物约10.7 g,产率92.0%;1H NMR(600 MHz,CDCl3)δ:5.12(d,J=3.1 Hz,1H),5.09(t,J=10.0 Hz,1H),4.59~4.49(m,2H),4.26(dd,J=12.5 Hz,2.2 Hz,1H),3.99(t,J=9.5 Hz,1H),3.80(ddd,J=9.5 Hz,3.9 Hz,2.2 Hz,1H),3.68(s,1H),3.52(s,3H),2.14(s,3H);13C NMR(151 MHz,CDCl3)δ:172.09,95.41,83.49,80.13,77.37,77.16,76.95,71.86,68.18,62.15,56.35,20.93。
(4) 甲基2,3-O-环磺酰基-4,6-乙酰氧基-α-D-吡喃葡萄糖苷(化合物Ⅳ)的合成
按化学计量比称量化合物Ⅱ(10.0 g,0.0390 mol)加入圆底烧瓶(250 mL)中,随后加入二氯甲烷约60 mL和6.6 mL的吡啶充分溶解后,再加入7.7 mL乙酸酐,在35 ℃下搅拌反应3 h,TLC监测反应完全后(展开剂:石油醚∶乙酸乙酯=7∶3,V∶V),向反应完全的混合体系中加入150 mL乙酸乙酯稀释,该有机相用水(2×50 mL)洗涤,分液,加入少量盐酸调节pH≈6,合并水相。水相再用50 mL乙酸乙酯反萃取,合并有机相,有机层用100 mL饱和食盐水洗涤,分液,得到有机相,经无水硫酸钠干燥,过滤,减压蒸馏得到白色固体12.5g,产率94.0%;1H NMR(600 MHz,CDCl3)δ:5.40(d,J=10.2 Hz,1H),5.17(m,2H),4.68(dd,J=10.2 Hz,3.0 Hz,1H),4.28(dd,J=12.6 Hz,4.8 Hz,1H),4.18(dd,J=12.6 Hz,2.4 Hz,1H),3.99~3.97(m,2H),3.69~3.66(m,1H),3.54(s,3H),2.13(s,3H),2.11(s,3H);13C NMR(150 MHz,CDCl3)δ:170.47,168.77,96.08,80.99,79.85,69.31,67.08,61.21,56.33,20.66,20.49。
(5) 甲基2,3-O-环磺酰基-6-O-苯甲酰-α-D-吡喃葡萄糖苷(化合物Ⅴ)的合成
按化学计量比称量化合物Ⅱ(10.0 g,0.0390 mol)加入圆底烧瓶(250 mL)中,随后加入二氯甲烷60 mL,再定量加入4.0 mL吡啶,冰浴下搅拌5 min混合均匀后加入5.8 mL苯甲酰氯,搅拌反应4 h,TLC监测反应完全后(展开剂:石油醚∶乙酸乙酯=7∶3,V∶V),向反应完全的混合体系中加入200 mL乙酸乙酯稀释,该有机相用水(2×50 mL)洗涤,分液,加入少量盐酸调节pH≈6.0,合并水相,水相再用50 mL乙酸乙酯反萃取,合并有机相,有机层用100 mL饱和食盐水洗涤,分液,得到有机相,经无水硫酸钠干燥,过滤,减压蒸馏得到白色固体12.8 g,产率91%;1H NMR(600 MHz,CDCl3)δ:8.10~7.94(m,2H),7.61(t,J=7.4 Hz,1H),7.46(t,J=7.6 Hz,2H),5.23~5.06(m,2H),4.89~4.77(m,1H),4.55(dd,J=10.4 Hz,3.1 Hz,1H),4.06(t,J=9.5 Hz,1H),3.95~3.88(m,1H),3.53(s,3H);13C NMR(150 MHz,CDCl3)δ:167.54,133.92,133.77,130.13,129.97,129.08,128.76,128.62,95.33,83.59,80.22,72.07,68.27,62.65,56.29。
(6) 甲基2,3-O-环磺酰基-4,6-O-二苯甲酰基-α-D-吡喃葡萄糖苷(化合物Ⅵ)的合成
按化学计量比称量化合物Ⅱ(10.0 g,0.0390 mol)加入圆底烧瓶(250 mL)中,随后加入二氯甲烷约60 mL,充分溶解后,再加入9.5 mL苯甲酰氯(或相应比例的者苯甲酸酐),室温下搅拌反应4 h,TLC监测反应完全后(展开剂:石油醚∶乙酸乙酯=7∶3,V∶V),向反应完全的混合体系中加入100 mL乙酸乙酯稀释,该有机相用水(2×50 mL)洗涤分液,加入少量盐酸调节pH≈6.0,合并水相,水相再用50 mL乙酸乙酯反萃取,合并有机相,有机层用100 mL饱和食盐水洗涤,分液,得到有机相,经无水硫酸钠干燥,过滤,减压蒸馏得到白色固体17.0 g,产率94.0%;1H NMR(600 MHz,CDCl3)δ:8.02(ddd,J=17.4 Hz,8.3 Hz,1.4 Hz,4H),7.65~7.53(m,2H),7.50~7.39(m,5H),5.78(t,J=9.7 Hz,1H),5.36(t,J=10.1 Hz,1H),5.21(d,J=3.1 Hz,1H),4.76(dd,J=10.3 Hz,3.1 Hz,1H),4.64(dd,J=12.4 Hz,2.8 Hz,1H),4.43(dd,J=12.3 Hz,4.8 Hz,1H),4.29(ddd,J=9.7 Hz,4.8 Hz,2.8 Hz,1H),3.58(s,3H);13C NMR(151 MHz,CDCl3)δ:166.09,164.57,134.20,133.52,130.16,129.80,129.41,128.78,128.63,128.26,95.24,81.15,80.07,69.64,68.25,62.19,56.52。
(7) 呋喃甲醛类衍生物的制备方法
按化学计量比任意称量上述化合物Ⅰ~Ⅵ的一种,加入圆底烧瓶中,随后加入至少5倍量的DMF,充分溶解后,再定量加入2倍当量的碳酸氢钠,在90 ℃下加热搅拌反应,TLC监测反应完全后,加入适量盐酸调节pH至中性,过滤,减压蒸馏和纯化得到目标产物。
(8) 5-HMF的合成
精馏(机械油泵(5~3 mmHg),收集:115~120 ℃)得到淡黄色固体,产率91.6%;1H NMR(600 MHz,CDCl3)δ:9.59(s,1H),7.22(d,J=3.5 Hz,1H),6.52(d,J=3.6 Hz,1H),4.72(s,2H);13C NMR(150 MHz,CDCl3)δ:177.81,160.63,152.54,110.13,57.81。
(9) 5-苯甲酰氧甲基糠醛的合成
柱层析(洗脱剂:乙酸乙酯∶石油醚=1∶10~1∶8,V∶V),淡黄色固体,产率87.6%;1H NMR(600 MHz,CDCl3)δ:9.65(s,1H),8.10~8.02(m,2H),7.58(tt,J=7.4 Hz,1.4 Hz,1H),7.44(t,J=7.8 Hz,2H),7.23(d,J=3.6 Hz,1H),6.67(d,J=3.6 Hz,1H),5.38(s,2H);13C NMR(151 MHz,CDCl3)δ:177.99,166.07,155.66,153.03,133.58,129.97,129.41,128.62,112.90,77.37,77.16,76.95,58.37。
(10) 5-乙酰氧甲基糠醛的合成
柱层析(洗脱剂:乙酸乙酯∶石油醚=1∶10~3∶7,V∶V),无色油状液体,产率90.0%;1H NMR(600 MHz,CDCl3)δ:9.63(s,1H),7.20(d,J=3.6 Hz,1H),6.58(d,J=3.6 Hz,1H),5.11(s,2H),2.10(s,3H);13C NMR(151 MHz,CDCl3)δ:177.83,170.33,155.44,152.87,112.59,77.25,77.04,76.82,57.81,20.70。
2 结果与讨论
首先参照Berridge的方法合成得到化合物Ⅰ[14](图3)。该化合物Ⅰ的立体结构非常独特,由于2-位和3-位的羟基处于吡喃糖环的相反位置,对于由离去基团构成的环硫酸酯结构而言,该5元环并6元环的叉向结构使得该离去基团产生离去作用,诱导生成2-位或3-位碳正离子是极其不利的,因此难以发生对应原子位点的亲核取代反应,例如该化合物Ⅰ在苯酚钾、醇钠或氢氧化钠等碱作用下,仅获得2,3-环氧化合物Ⅶ[14],其扭曲的叉向环硫酸酯结构使得该吡喃糖环结构具有极强的分子内张力,从而诱导出一些独特的化学性质。实验研究发现化合物Ⅰ在含水溶剂条件下,即使无酸性催化剂的存在,在室温下亦可缓慢脱除4-和6-位的丙叉保护基,生成化合物Ⅱ。化合物Ⅱ与具有位阻效用的酰化试剂进行酰化反应时,具备较好的选择性,如当使用1 eq.的苯甲酰氯和吡啶进行酰化时,室温下仅获得化合物Ⅴ;相同条件下,替换乙酰氯作为酰化试剂时,则以1∶3的物质的量之比获得化合物Ⅲ和化合物Ⅳ。进一步化合物Ⅱ溶解在冰乙酸中,滴入催化量的浓硫酸,仅获得化合物Ⅲ。
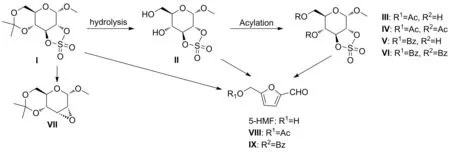
图3 5-HMF的合成路线
为了进一步探索该环硫酸叉酯结构对吡喃糖环的化学性质的影响,直接采用化合物Ⅰ在各种溶剂下进行加热分解,当温度达到80 ℃时,发生缓慢分解,经过提取分离纯化,获得的主要产物为5-HMF。进一步优化该反应条件,利用碱中和反应过程中生成硫酸,该结构以高达90%以上的收率获得目标产物。在相同反应条件下,分别利用化合物Ⅰ~Ⅵ进行反应,均获得对应的5-羟甲基具有不同保护基的5-HMF的衍生物,所有这些均为以D-葡萄糖为单体,高效制备5-HMF系列衍生物提供很好的方法学支持。
3 结论
以廉价的生物质衍生物——无水D-葡萄糖为起始原料制备5-HMF的最简生产流程为:按照MATTHIAS[15]和BAZIN[16]方法,先后进行甲苷化和丙叉化反应获得甲基4,6-亚丙基-α-D-吡喃葡萄糖苷,2步反应经后处理直接获得产物,均不需要进一步的纯化即可以直接用于后一步的反应,2步反应的总产率为90%。随后该中间体经过环硫酸叉酯化获得化合物Ⅰ,无需进一步的纯化最终经过高温脱水获得5-HMF,该制备路线的总产率可达61.8%(0.9%×0.75%×0.916%)。虽然该方法是通过多步反应的形式进行,但是由于每一步的产率相对比较高,在获取到最终目标物之前,实验证明均不需要进行额外的分离纯化,所有此过程中使用的试剂均不存在剧毒,易爆和难以回收等不利生产的环境和安全问题,最终产品可以采用精馏的形式获得,因此该新工艺路线具备极强的新颖性和创造性,且符合现有绿色工业化的特点,为5-HMF的制备提供一个很好的方法选择。
猜你喜欢
净水技术(2022年11期)2022-11-10
化学工程师(2022年5期)2022-05-11
中国纤检(2021年9期)2021-09-30
中国特种设备安全(2021年2期)2021-07-21
石油地质与工程(2019年3期)2019-09-10
中国钼业(2019年2期)2019-01-19
水利技术监督(2016年6期)2017-01-15
当代化工(2016年9期)2016-10-28
合成化学(2015年9期)2016-01-17
烟草科技(2015年8期)2015-12-20
