维胺酯原料药的有关物质分析
2024-03-14 03:48李钰鑫兰文刘雁鸣湖南省药品检验检测研究院长沙410001国家药品监督管理局药用辅料工程技术研究重点实验室长沙410001
中南药学 2024年2期
李钰鑫,兰文*,刘雁鸣(1.湖南省药品检验检测研究院,长沙 410001;2.国家药品监督管理局药用辅料工程技术研究重点实验室,长沙 410001)
维胺酯,化学名为N-(4-乙氧碳基苯基)维甲酰胺,因结构与全反式维A 酸类似,其作用机制与13-顺维A 酸及芳香维A 酸较相似,但刺激性低于维A酸,不良反应比维A酸少,治疗指数为维A酸的10倍[1-2]。维胺酯能够调节和控制上皮细胞分化与生长,减少皮脂分泌,抑制角质形成细胞的角化过程,使角化异常恢复正常,发挥抗雄激素、抗炎、调节免疫及预防瘢痕形成等作用,用于治疗各种痤疮、角化异常性皮肤病等,同时对鱼鳞病、银屑病(牛皮癣)、苔藓类皮肤病、黏膜病及结缔组织等疾病也有一定疗效[3-5]。维胺酯为我国自行研制的维A酸类药物,维A酸作为起始原料与三氯化磷反应转化为维A酰氯,再与苯佐卡因反应生成维胺酯,反应路线如图1所示。目前国内仅3家维胺酯原料生产企业,国内外药典均未收载该品种[6-7],现行标准为《国家药品标准》化学药品地标升国标第二册[WS-10001-(HD-0164)-2002]和原国家食品药品监督管理总局标准YBH14602008,与《国家药品标准》化学药品地标升国标第二册标准比较,原国家食品药品监督管理总局标准YBH14602008增订了有关物质及残留溶剂检查,有关物质方法仅对杂质总量进行控制。本文在现行标准基础上,结合《中国药典》2020年版四部指导原则[8],首次建立了同时测定维胺酯中维A酸和其他杂质的HPLC方法;并采用液质联用技术[9-10]结合强制降解试验,对主要未知杂质峰进行定性分析并推测杂质来源。采用ADMET Predictor 软件[11]对维胺酯及其主要杂质进行毒性预测,为制订维胺酯原料药的质量标准提供参考。
1 材料
Ultimate3000型高效液相色谱仪(美国戴安公司);十万分之一MS205DU电子天平(瑞士梅特勒-托利多公司);1290-6550型高效液相色谱-四极杆-飞行时间质谱(美国安捷伦公司)。
维胺酯对照品(纯度:99.03%,批号:190303,C公司,湖南省药品检验研究院标定);维A酸对照品(纯度:99.4%,批号:100307-200902,中国食品药品检定研究院);甲醇(色谱纯,Burdick &Jackson);水为纯化水;其他试剂均为分析纯;15批维胺酯原料药(A公司,批号分别为170101、170102、170103、181002;B公司,批号分别为161205、161206、190303、190304、190305;C公司,批号分别为V2A20160501、V2A20160502、V2A20160503、VIA-20190401、VIA-20190402、VIA-20190403)。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Eclipse XDB C18(4.6 mm×250 mm,5 μm);流动相:甲醇-水(93∶7);流速:1.0 mL·min-1,检测波长:370 nm;进样量:20 μL。
2.2 溶液的制备
2.2.1 供试品溶液的制备 取维胺酯约25 mg,精密称定,置50 mL量瓶中,加甲醇适量超声使溶解,用甲醇稀释至刻度,摇匀,滤过,取续滤液即得。
2.2.2 对照品溶液的制备 精密称取维A酸对照品约12.5 mg,置100 mL量瓶中,用甲醇适量超声溶解并稀释至刻度,摇匀,精密量取1 mL,置50 mL量瓶中,用甲醇稀释至刻度,摇匀,即得。
2.2.3 对照溶液的制备 精密量取供试品溶液1 mL,置100 mL量瓶中,加甲醇稀释至刻度,摇匀,即得。
2.2.4 系统适用性溶液的制备 取维胺酯对照品适量,加甲醇超声溶解并稀释成每1 mL中约0.5 mg的溶液,取溶液适量,于(2500±500)lx光照强度下照射30 min,即得。
2.3 方法学验证
2.3.1 专属性 取“2.2”项下系统适用性溶液、对照品溶液、供试品溶液及稀释剂(甲醇)各20 μL进样测定,结果显示维胺酯的出峰时间约15.6 min,维A酸的出峰时间约6.363 min,峰形对称性较好,维A酸理论塔板数在11 000以上,主峰与已知杂质维A酸及其他未知杂质峰均有效分离。空白溶剂对测定无干扰,结果见图2。
2.3.2 强制降解试验 采用光照、酸、碱、氧化、高温等剧烈条件对本品(批号:170101)进行破坏性降解处理。取本品25 mg,平行5份,分置50 mL量瓶中,经以下条件进行破坏处理:① 光照破坏[于照度(2500±500)lx下放置5 d];② 酸破坏(1 mol·L-1盐酸溶液0.5 mL,放置4 h,加5 mol·L-1氢氧化钠溶液调至中性);③ 碱破坏(1 mol·L-1氢氧化钠溶液0.5 mL,放置4 h,加5 mol·L-1盐酸溶液调至中性);④ 氧化破坏(3%过氧化氢溶液1 mL,放置4 h);⑤ 高温破坏(置150℃烤箱中放置4 h,放冷)。结果表明:光照破坏试验中,在主峰相对保留时间0.9处的未知杂质峰面积明显增加,命名该杂质为杂质A,说明本品易被光破坏,后期试验均采用避光操作;其他破坏试验产生了少量杂质,各杂质均能与主峰完全分离,表明本色谱系统专属性良好。在各破坏试验记录的色谱图中,主峰保留时间2倍以后的基线平稳且没有检测出其他杂质峰,故供试品溶液的采集时间拟定为主峰保留时间的2倍,见图3。
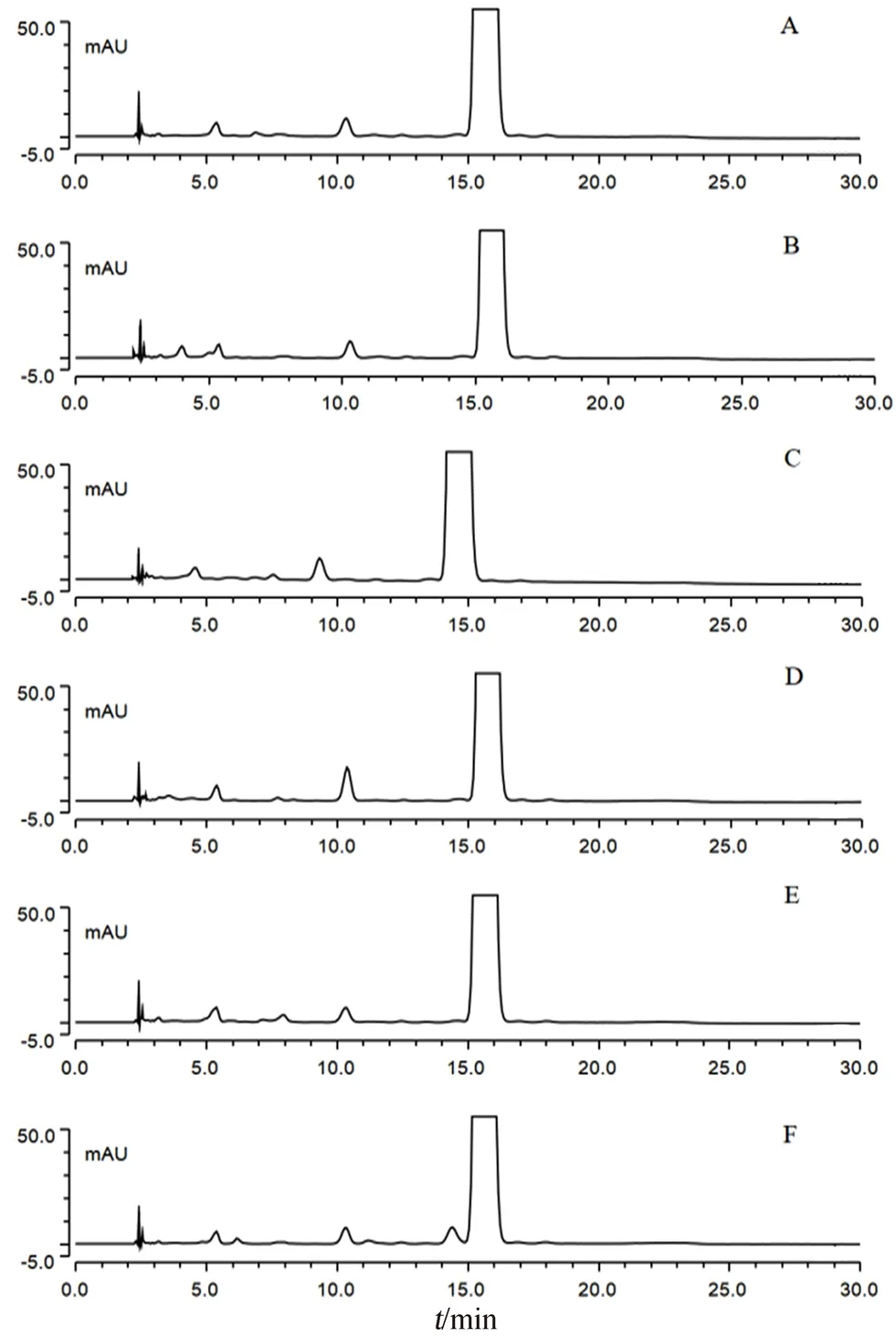
图3 维胺酯强制降解试验色谱图Fig 3 HPLC chormatogram for forced degradation test of viaminate
2.3.3 维A酸的线性关系考察 取维A酸对照品20.43 mg,置100 mL棕色量瓶中,加甲醇超声溶解并稀释至刻度,摇匀,作为维A酸对照品储备液。分别精密量取维A酸对照品储备液适量,用甲醇依次稀释配成质量浓度约为0.1、0.5、1.0、2.5、5.0、10.0、50.0 μg·mL-1的系列对照品溶液。精密量取上述溶液各20 μL,依法测定,以质量浓度(X)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线,得回归方程:Y=136.34X-15.255,r=1.000,结果表明,维A酸在0.1015~50.76 μg·mL-1与峰面积线性关系良好。
2.3.4 进样精密度试验 取“2.3.3”项下第4份溶液,精密量取20 μL,注入液相色谱仪,连续进样6次,记录色谱图,结果RSD为0.67%,表明仪器精密度良好。
2.3.5 回收试验 精密称取已知维A酸杂质量(0.052%)的样品(批号:170101)约25 mg 6份,分别置50 mL棕色量瓶中,各精密加入维A酸对照品溶液(质量浓度为0.1244 mg·mL-1)1 mL,加甲醇溶解并稀释至刻度,摇匀,作为供试品溶液,按“2.1”项下色谱条件进样测定并计算回收率,平均回收率为98.12%,RSD为0.53%。
2.3.6 定量限及检测限考察 取维胺酯和维A酸对照品各适量,逐级稀释,各取20 μL,注入液相色谱仪,记录色谱图。设定量限S/N=10,检测限S/N=3,得维胺酯和维A酸的定量限依次为0.058 μg·mL-1和0.074 μg·mL-1,检测限依次为0.020 μg·mL-1和0.039 μg·mL-1,
2.3.7 重复性试验 取“2.3.5”项下6份供试品溶液20 μL,按“2.1”项下色谱条件进样测定,测得维A酸的量为0.59%,RSD为1.0%;其他单个杂质量为0.28%,RSD为1.0%;各杂质总量为1.0%,RSD为1.2%。
2.3.8 稳定性试验 取“2.2”项下供试品溶液,分别于室温放置0、2、4、8、12、24 h后,进样测定,结果供试品溶液中维A酸、其他单个杂质及各杂质的总峰面积的RSD分别为1.3%、0.88%及0.60%,表明供试品溶液在24 h内稳定。
2.3.9 耐用性试验 使用3个品牌色谱柱[Agilent Eclipse XDB C18(4.6 mm×250 mm,5 μm)、Welch Ultimate X C18(4.6 mm×250 mm,5 μm)、CAPCELL TYPE MGⅡ(4.6 mm×250 mm,5 μm)]调整柱温、流速、流动相比例等色谱条件进行耐用性考察,结果峰形和分离均较好,精密称取维胺酯(批号:170101)约25 mg 依法测定有关物质,结果见表1。
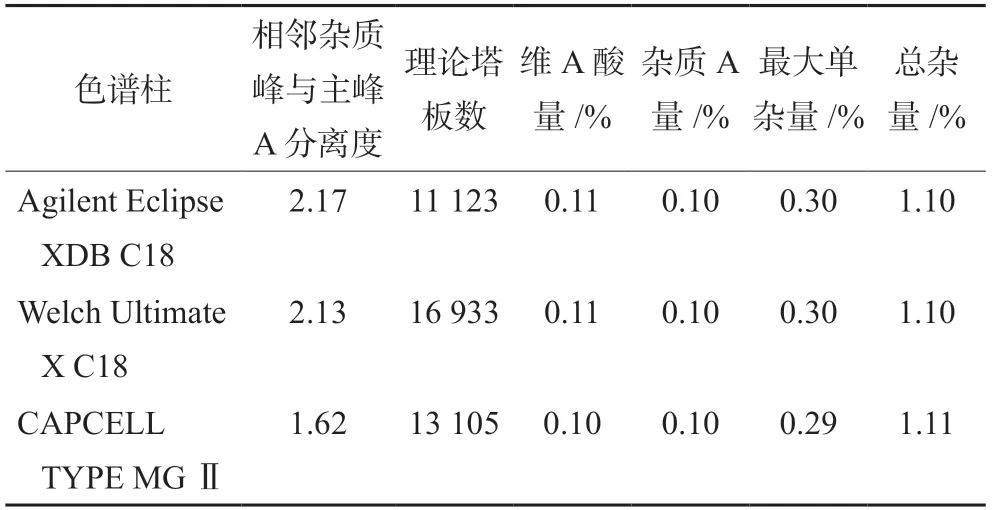
表1 维A酸的耐用性试验结果Tab 1 Durability of retinoic acid
2.4 样品含量的测定
取3家公司的15批样品依法测定有关物质,维A酸含量以外标法按峰面积计算,其他杂质含量采用主成分自身对照法计算,结果见表2。
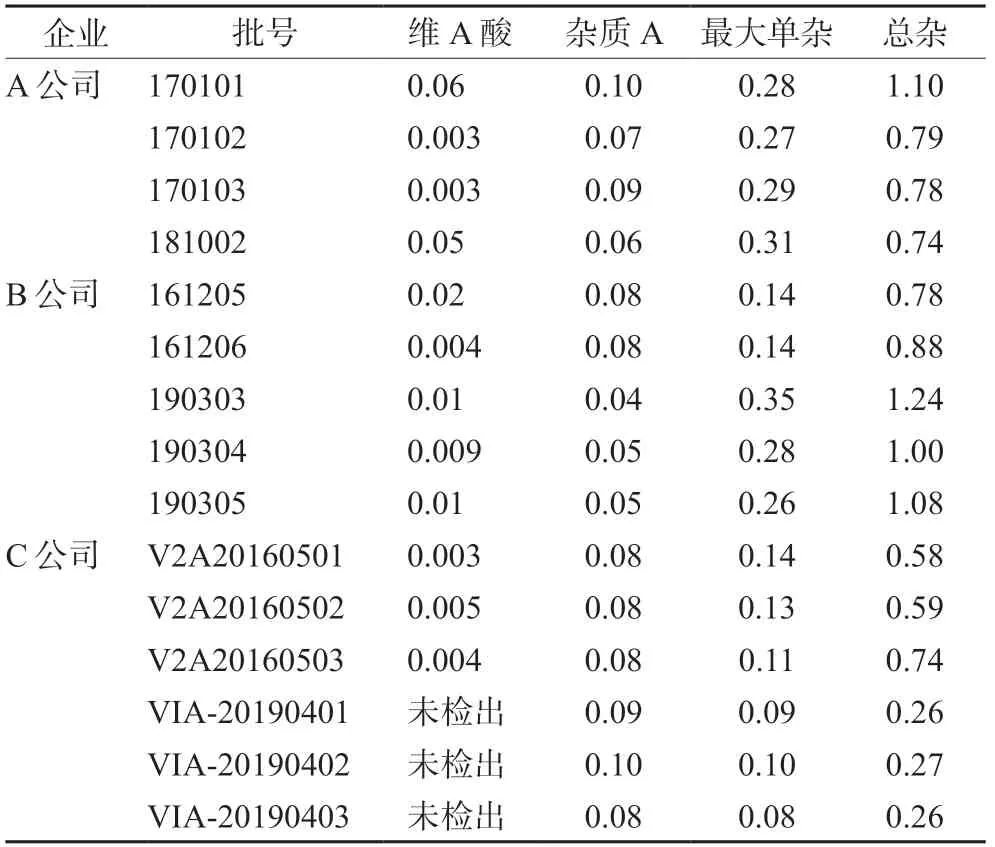
表2 样品有关物质测定结果(%)Tab 2 Related substances of samples (%)
2.5 未知杂质质谱分析
样品的测定结果表明,3家公司的15批样品均检出降解杂质A,且含量达0.1%,可能存在安全隐患,有必要对未知杂质进行归属。本文采用高效液相色谱-四极杆-飞行时间液质联用仪[10],在ESI负离子模式下,得到该未知杂质的一、二级质谱信息,结果显示杂质A与维胺酯的分子离子峰和离子碎片几乎相同,结果见图4。
2.6 杂质毒性预测和评价
使用Chemdraw软件画出维胺酯及杂质的结构式并保存为“.mol”格式,导入ADMET Predictor软件,对维胺酯及维A酸、杂质A的毒理性质参数进行全面预测。结果显示三者主要毒性为肝脏毒性,另外还有潜在致敏性和生殖毒性,维A酸的预测毒性低于维胺酯,杂质A与维胺酯的毒性预测结果基本一致,结果见表3。

表3 ADMET Predictor 软件预测结果Tab 3 Predict outcomes by ADMET Predictor software
3 讨论
3.1 色谱柱的选择
维胺酯脂溶性较强,且遇光易变质,产生的降解杂质与主峰较难达到基线分离,故选择光破坏溶液作为系统适用性溶液。现行标准YBH14602008有关物质检查方法采用C18柱,采用不同品牌、不同型号的C18柱,适当调整流动相比例,均能将维胺酯与光降解杂质有效分离。
3.2 溶剂的选择
分别考察用流动相、甲醇及水作为溶剂时对有关物质测定的影响,结果显示当使用水为溶剂时,样品几乎不溶解;当使用流动相为溶剂时,样品很难溶解。最终选择甲醇作为稀释剂,供试品溶解完全,且稀释剂与主成分维胺酯对待测组分无干扰。
3.3 波长的选择
截取系统适用性溶液DAD图中维胺酯的紫外光谱图、对照品溶液DAD图中维A酸的紫外光谱图、供试品溶液DAD图中主要杂质的紫外光谱图及破坏试验DAD图中主要降解杂质的紫外光谱图,结果显示:维胺酯在370 nm波长处有最大吸收,特殊杂质维A酸、杂质A及其他主要杂质在该波长处都有一定的吸收,故选择370 nm作为有关物质的检测波长。
3.4 杂质A的来源分析
强制降解试验表明,维胺酯光照敏感,在强光条件下降解产物杂质A的峰面积明显增加,进一步对该杂质进行质谱分析,其质谱图与维胺酯质谱图一致,经企业调研和文献查阅,维胺酯为全反式维A酸的衍生物,其为全反式维A酸的酸根经酰化后,与氨基反应生成的衍生物,因此也为全反式结构,且维A酸见光易转化为其顺式异构体异维A酸,推断其为维胺酯的同分异构体(或顺式维胺酯)。杂质A来源分析见图5。

图5 杂质A可能来源Fig 5 Formation mechanism of impurity A
3.5 限度拟订
维A酸为维胺酯合成的起始原料,直接反映维胺酯生产工艺的优劣;杂质A为维胺酯的主要降解杂质,反映其储存条件下质量的优劣,故对杂质维A酸及杂质A进行质量控制。现行标准有关物质项中仅对总杂质量进行控制,限度为2.0%;参考ADMET Predictor软件毒性分析结果:维A酸、杂质A同维胺酯的毒性相当;同时考虑维胺酯相关制剂的治疗周期为4~6周,非长期用药;结合各企业近效期样品的试验考察结果以及工艺控制情况,暂拟订限度为维A酸不得过0.5%,杂质A不得过0.5%,其他单杂不得过0.5%,各杂质总和不得过2.0%。
3.6 维胺酯的质量现状
根据对国产3家主要生产企业产品的有关物质检查结果,工艺杂质维A酸含量均小于0.1%,说明国内企业对该杂质控制均较好;各企业均检出光降解杂质A,说明在样品储存过程中,企业应严格控制储存条件,避免其光照降解。
4 结论
本文建立了一种有效检测维胺酯原料药中有关物质的高效液相色谱法。基于质谱数据,结合强制降解试验结果以及维胺酯的处方和生产工艺,推测维胺酯中主要杂质的结构与来源,并用计算机软件预测杂质的毒性,为维胺酯的质量控制奠定了基础,为该品种的处方和生产工艺、质量控制及储存条件提供了参考依据。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
艺术品鉴(2020年6期)2020-12-06
中成药(2018年12期)2018-12-29
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
Asian Journal of Urology(2015年3期)2015-12-16
中学生数理化·中考版(2015年12期)2015-09-10
江西化工(2015年5期)2015-04-17