
3 例新生儿期起病的血友病A 临床特点及基因分析报道
2024-03-11 03:54黄玲玲王宁玲储金华涂松济吴正玉
安徽医学 2024年2期
黄玲玲 王宁玲 储金华 涂松济 吴正玉
1 病例资料
患儿1:男性,9 d,因“皮肤黄染1 周”入院,查体:重度黄染,头顶部可触及10 cm×6 cm 血肿,右腋下及上臂静脉穿刺处血肿。实验室检查示活化部分凝血活酶时间(activated partial thromboplastin time,APTT)164.9 s(正常参考值25~31.3 s),凝血因子活性检测示凝血因子Ⅷ活性0.9%(正常参考值50.0%~150.0%),凝血因子Ⅱ、Ⅶ、Ⅸ、Ⅹ及血管性血友病因子(von willebrand factor,VWF)均正常。凝血因子Ⅷ内含子22 及内含子1正常,二代测序(2021.11)在FⅧ基因Exon8 上检出无义变异c.1063C>T(p.R355*)(半合),患儿母亲携带无义变异c.1063C>T(p.R355*)(杂合)。确诊为血友病A 型(Hemophilia A,HA)。
患儿2:男性,1 h 55 min,因“胎龄(35+6)周,生后反应差24 min”入院,住院期间出现黄疸及右上肢肘窝静脉穿刺处血肿、双侧手腕静脉穿刺处血肿,实验室检查示APTT 116.9 s,凝血因子Ⅷ活性1%,凝血因子Ⅱ、Ⅶ、Ⅸ、Ⅹ及VWF 正常。肘关节超声示右肘内侧肌层内低回声(考虑血肿),腕关节超声示左腕部皮下无回声(考虑血肿)。凝血因子Ⅷ内含子22 及内含子1 正常,二代测序(2021.12)在FⅧ基因Exon4 上检出错义变异c.464A>G(p.Y155C)(半合),据目前数据库检索,未见报道,暂不明确病理意义,但HGMD 中收录变异位点 c.464A>C(p.Y155S)为病理性位点;在Exon14 上检出错义变异 c.4531G>A(p.V1511I)(半合),据目前数据库检索,该变异为HA 相关的病理性位点(CM082634) <PMID:33706050,PMID28056528>。患儿母亲同时携带c. 464A>G(p. Y155C)(杂合)和c. 4531G>A(p. V1511I)(杂合)。确诊为HA。
患儿3:男性,1 个月25 天,因“反复多部位出血1 月余”入院,期间曾有黄疸及抽搐发作,头颅MRI 提示:①考虑双侧大脑半球大面积脑梗死;②考虑双侧颞顶部枕部硬膜下出血。住院期间右侧腹股沟抽血处出现皮下血肿,治疗期间多次监测APTT明显延长。实验室检查示 APTT 99.7s、凝血因子Ⅷ活性0.53%,凝血因子Ⅸ、VWF 正常。超声检查示双侧肘关节及膝关节未见异常、右侧腹股沟低回声包块(考虑血肿)。凝血因子Ⅷ内含子22 及内含子1 正常,二代测序(2023.04)示FⅧExon14无义变异c. 2294C>G(p. S765X)(半合),见图1。患儿母亲携带c.2294C>G(p.S765X)(杂合),见图2。确诊为HA。
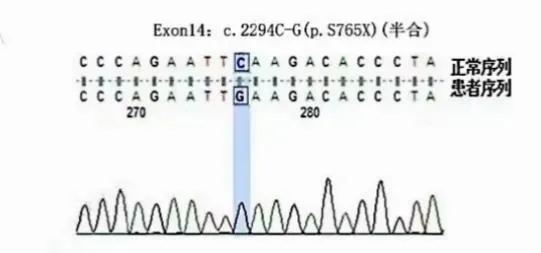
图1 患儿c.2294C>G半合突变
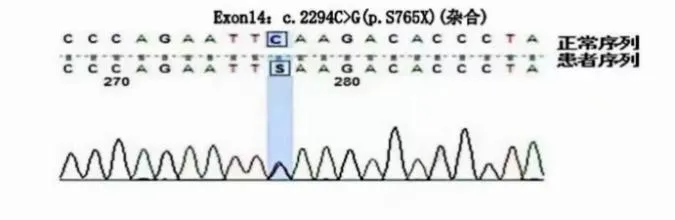
图2 患儿母亲c.2294C>G杂合突变
2 讨论
HA(OMIM:306700) 是一种凝血因子Ⅷ缺陷引起的X 染色体连锁隐性遗传性出血性疾病。FⅧ基因位于X 染色体长臂末端(Xq28),长度超过180 kb,结构相当复杂,包含26 个外显子[1]。FⅧ基因的突变位点遍布整个基因,截至2020 年2 月已有4 344 种FⅧ基因突变被收录在人类基因突变数据库(human genetic mutation database,HGMD)中。这些突变类型多样,包括内含子22 及1 倒位突变、大片段缺失/插入、无义突变、错义突变、剪接位点突变、小片段缺失/插入、调控区域突变、复杂重排等,其中点突变为最常见的基因缺陷,存在于约90%的HA 患者中。本研究报道了3 例新生儿或婴儿期的HA 患儿,这些患儿均携带FⅧ基因的点突变,并且这些突变是通过母亲遗传给患儿。值得注意的是,病例3 中的无义变异p.S765X 是首次报道。
HA 的临床表现为延迟、持续而缓慢的渗血,出血频度与部位取决于患者体内的凝血因子水平,根据FⅧ的水平HA 分为三型,即轻、中、重型。重型患儿出血表现明显,而轻型患儿仅表现为外伤及手术后的出血倾向[2]。一项11 例新生儿HA 研究显示,新生儿中重型HA 并发颅内出血发生率高,同时部分患儿存在黄疸出现时间早、程度重[3]。本研究中3 例患儿均出现黄疸及皮肤血肿,其中患儿1 及患儿3 为重型,患儿3 病程长,期间发生重要脏器出血(颅内出血),患儿1 早期明确诊断,未发生重要脏器出血。新生儿HA 合并症和死亡的主要原因为颅内出血[3],早期诊治可改善预后。
HA 的基因突变通常与其表型相关,这是由于不同的突变对FⅧ蛋白活性产生了不同的效应。内含子22 及1 倒位、大片段缺失、无义突变常引起FⅧ蛋白大部分缺失从而导致重型血友病A,错义突变可引起个别氨基酸替换从而导致轻/中型HA。患儿1 和患儿3 的无义突变产生缩短的蛋白质分子,通常被信使RNA 的无义介导途径(nonsense-mediated mRNA decay,NMD)[4]所识别和降解,因此会导致凝血因子Ⅷ功能的丧失或极度缺乏,进而引起重型HA 的表型。患儿3 的p. S765X 经NCBI/HGMD/ClinVar/HAMSTeRS /SNP 等数据库检索未见报道,但根据ACMG 分类规则为HA 致病变异,系新的突变。患儿2同时检出p. Y155C 和p. V1511I 这2 个错义突变,错义突变引起HA 的严重性取决于替换的氨基酸和它的位置。Y155 位于蛋白A1 亚基,而A1 亚基与蛋白质合成及其活化密切相关[5],p.Y155C 可能导致残基的大小和亲水性发生改变,进而可能降低蛋白质与Ca2+金属离子的相互作用能力,导致FⅧ活性降低,经EAHADF8基因变异数据库检索,一例HA 患者中存在p.Y155C突变,分型为重型。V1511 位于蛋白B 亚基的未定义区域,B 亚基通过调节FⅧ蛋白的分泌影响其活性, B 亚基的错义突变多数可能为基因多态性或非致病性突变,而且B 亚基的错义突变多数不会导致重型HA[6]。检索发现仅在1 例台湾重型HA 患者中同时存在内含子22 倒位和p. V1511I[7],并且PolyPhen-2与Mutation Taster 多个软件预测p. V1511I 变异无致病性。因此,患儿2 的HA 致病突变为p.Y155C。
凝血因子替代治疗是HA 急性出血时最有效的止血措施,预防治疗是儿童HA 治疗的首选方法,重型患儿应及早开始预防治疗,治疗过程中应监测HA 抑制物并给与相应治疗[2]。需注意在重型HA 患者中,无义突变者较内含子22 倒位者具有更高的FⅧ抑制物产生风险[8]。本研究中患儿1 和患儿3 为重型、无义突变,均有出血表现,给与替代治疗和预防治疗,随访期间无出血表现,监测抑制物阴性。
综上,随着对HA 认识的提高及新突变位点的发现,HA 基因突变数据库逐步完善。本病例报道总结了新生儿HA 相关临床特点,加强临床对该病的认识,并报告1 例FⅧ基因突变新位点,丰富了FⅧ基因突变数据库。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30
北京大学学报(医学版)(2022年2期)2022-11-21
昆明医科大学学报(2022年3期)2022-04-19
中国当代儿科杂志(2021年6期)2021-06-16
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
中南医学科学杂志(2019年6期)2019-12-05
生物工程学报(2019年6期)2019-07-10
遗传(2019年5期)2019-05-21
生物学通报(2019年1期)2019-02-15
岷峨诗稿(2018年2期)2018-11-14
