
硝酸钍法测定排渣中氟含量的方法改进研究
2024-03-04 09:22金文进王若钦
当代化工研究 2024年3期
*金文进 王若钦
(1.甘肃工业职业技术学院 化工学院 甘肃 741025
2.白银中天化工有限责任公司 甘肃 730621)
化工生产中氟的含量分析通常有五种方法:电感耦合等离子体原子发射光谱法、F离子检测计法、EDTA滴定法、X射线荧光光谱法及硝酸钍滴定法。目前常用的是硝酸钍滴定法,它能较为快速的测出氟化钙的含量,而且操作较为简单,常被许多生产企业的化验室所采纳。但该法也有一些缺陷:采用的是指示剂控制终点,而人眼对色彩的辨识因人而异会出现偏差,导致滴定终点变色不明显,结果误差较大。同时,其中部分步骤流程中概括性语言较多,有些步骤描述比较笼统,没有细化,实际操作过程中分析人员不好把控,常常会根据自己的理解和习惯进行处理,导致相同样品同一实验室不同人员分析处理的结果不同,甚至差异较大,而且操作规程版本多,好多步骤都没有完全统一要求,这给实际的化工生产带来了一定挑战。为解决此法存在的缺陷和不足,优化及改进硝酸钍滴定法测定中的关键环节,细化操作,明确操作要领,确定操作规程中与分析结果准确度有关的因素,本试验以X射线荧光光谱法为对照,对样品前期处理、称量克重控制、溶样中氯离子是否洗干净及酸度调节是否到位等方面分别进行试验验证[1-2],以期搞清楚硝酸钍滴定法测定氟化钙含量的一些关键控制环节,为氟化工生产及实际应用提供一定的帮助。
1.材料和方法
(1)材料
排渣试样来自白银中天化工有限责任公司一车间,无明显颗粒物。首先是烘干样品,其次是样品脱水,干燥,冷却后将样品放入矿石研磨机中,所有的样品磨样控制磨样时间在0.5min,试样粒度研磨至75μm以下,进行以下实验。
(2)试剂
①标准试剂
A.氢氧化钠标准溶液c(NaOH)=0.1mol/L
配制:称取110g氢氧化钠,溶于100mL无二氧化碳的水中,摇匀,注入聚乙烯容器中,密闭放置。用塑料管量取上层清液5.4mL,用无二氧化碳的水稀释至1000mL,摇匀。贴上标签,待测定。
标定:准确称取在105~110℃电烘箱中干燥至恒重的工作基准试剂邻苯二甲酸氢钾约0.7000g,于250mL锥形瓶中,加无二氧化碳的水溶解,加2滴酚酞指示液,用配制好的氢氧化钠溶液滴定至溶液呈粉红色,并保持30s。记下氢氧化钠溶液消耗的体积,平行测定3次,同时做空白试验。
式中,
c(NaOH)—NaOH标准滴定溶液的浓度,mol/L;
m(KHC8H4O4)—领苯二甲酸氢钾的质量,g;
M(KHC8H4O4)—领苯二甲酸氢钾的摩尔质量,g/mol;
V(NaOH)—滴定时消耗NaOH标准滴定溶液的体积,mL。
分取已配制的0.1mol/L氢氧化钠10mL,用水稀释至100mL使用,必要时进行重新标定。
B.硝酸钍标准溶液T(Tn(NO3)4)=0.25mgF/mL
配制:称4.5000g四水合硝酸钍[Th(NO3)4·4H2O]于烧杯中,加水溶解后,移入5L试剂瓶中,冲至5L,摇匀,次日标定。
标定:称取在600℃灼烧1h的基准氟化钠0.2500g于烧杯中,加少量水加热使其溶解,待冷却后,转移入200mL容量瓶中,冲至刻度,摇匀备用。移取上述溶液10.00mL于三角瓶中,加水50mL,0.5mL茜素红(0.5g/L)用盐酸(0.06mol/L)调到溶液呈黄色,加入3mL茜素红(0.5g/L)后,再加入1mL醋酸缓冲溶液(1mol/L)调到pH在3.4±0.1,加入0.5mL次甲基溶液(0.5g/L)。用硝酸钍标准溶液滴定到刚刚出现蓝紫色为终点。硝酸钍标准溶液的浓度按式(2)计算。
式中:
T(Th(N03)4)—硝酸钍标准溶液的浓度,mgF/mL;
m—称取基准氟化钠的重量,g;
V—标定时消耗硝酸钍标准溶液的体积,mL;
0.4525—1克氟化钠相当于氟的克数。
②非标试剂
盐酸(AR):0.06mol/L;次甲基蓝AR 0.1%;茜素红:0.5g/L。溴化钾(AR)等。
(3)主要仪器
荧光光谱仪,坩埚,熔样炉TNO2C,酸碱滴定仪,锥形瓶,可控温电炉,pH计。
(4)方法
荧光光谱仪法[3]:精准称量2.0000g混合熔剂于50mL铂黄金皿(95% Pt+5% Au)中,再在熔剂上精准加入样品0.5000g,再在样品上盖上混合熔剂,加入几滴脱模剂(跟做曲线的滴数相同,跟铂黄金锅的状态有关)、10%溴化钾于坩埚中,于970℃使用熔样炉TNO2C,熔融样品10min。熔融结束后,待样品冷却,使样品在坩埚中自流凝固形成玻璃样片,样片应当坩埚底部一面光滑,样品呈均匀圆形,无气泡,样片无裂缝,边缘光滑,测样:将冷却好的样品装入样品盒,校准高度后,点样品,定量分析。
硝酸钍滴定法[4-5]:称(1.0000±0.0020)g排渣样置于锥形瓶中,加水约80mL放电炉上煮沸,然后加入25mL 1:1的盐酸继续加热煮沸至澄清,用定量滤纸过滤至500mL容量瓶中,用热水洗涤5~7次,用硝酸银法[3]鉴别是否将盐酸中Cl-清洗干净,定容摇匀,移取25.00mL试样,里面加茜素红8滴,再用6mol的氢氧化钠调微红,再用0.06mol/L盐酸调至无色,检测其pH在3.2~3.4之间,再加1mL茜素红试剂,1mL 1mol/L的醋酸溶液,1滴次甲基蓝指示剂,用硝酸钍滴定到溶液为蓝黑色即为滴定终点[4]。过滤的滤纸放入恒重过的瓷坩埚中,每位检验员各进行四次测定,并取其平均值,同时做空白试验。
氟化钙含量计算公式如下:
式中,C—硝酸钍标准滴定溶液浓度,mol/L;V—滴定试样耗费硝酸钍标准滴定溶液体积,mL;m—样品质量,mL;G—灰化后不溶物的质量分数,%。
检验人员:共设计了4位检验人员对每一步检验都同时采用两种方法进行测定。
2.结果与讨论
(1)样品粒度及称重对结果的影响
为了明确硝酸钍法中样品粒度和称重是否会影响氟化钙的含量,试验按照规程操作要求认真对样品首先进行了统一研磨,由4个化验员对同一样品各自取样后分别进行X射线荧光光谱法和硝酸钍滴定法进行氟化钙含量的测定,当称取质量在(1.0000±0.0020)g时,两种方法检测结果如表1所示。结果表明,X射线荧光光谱法测定的结果数据较为一致,而硝酸钍法结果差异较大。这说明该样品粒度处理及称重这一实验环节不是影响氟含量准确度的重要因素。
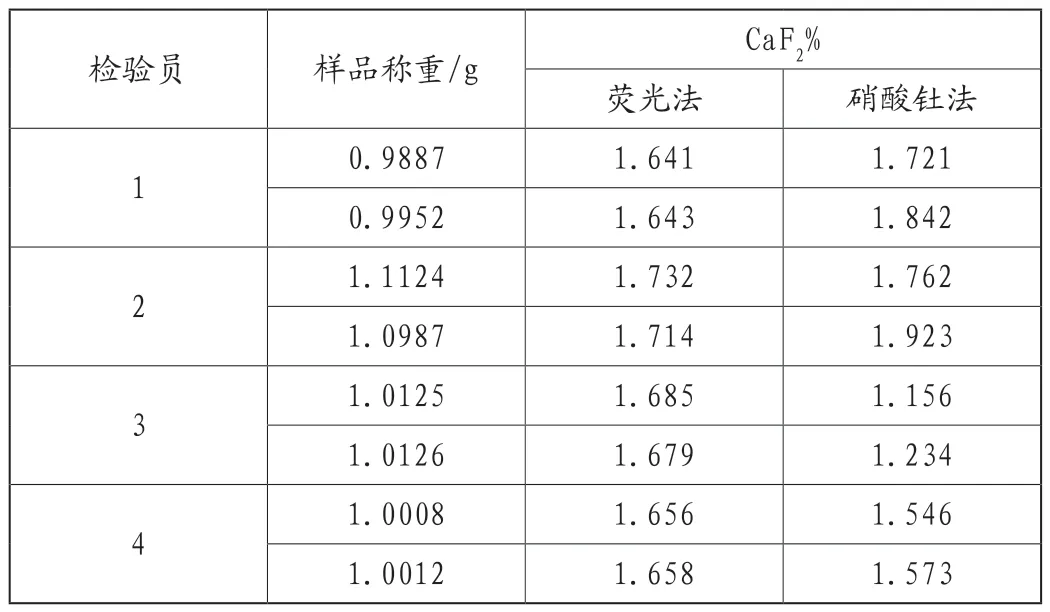
表1 样品粒度及称重对结果的影响
(2)溶样中酸度对结果的影响
按照硝酸钍滴定法的操作规程对分析检测中溶样酸度是否干扰测定结果的准确度进行了试验验证。将样品过滤时用热水统一洗涤9~10遍,再用硝酸银法检测样品中溶样盐酸是否洗干净,结果显示无沉淀生成,说明酸度洗涤这一步完全合适,然后再按照正常的荧光法和硝酸钍测定氟含量,结果如表2所示。可以看出,同一样品,处理方法相同,但4个化验员采用X射线荧光光谱法结果在误差范围内,而采用硝酸钍法测定结果差异仍比较大,这说明规程中溶样酸度在日常处理中正常,它也不是影响硝酸钍测定结果准确度的关键因素。
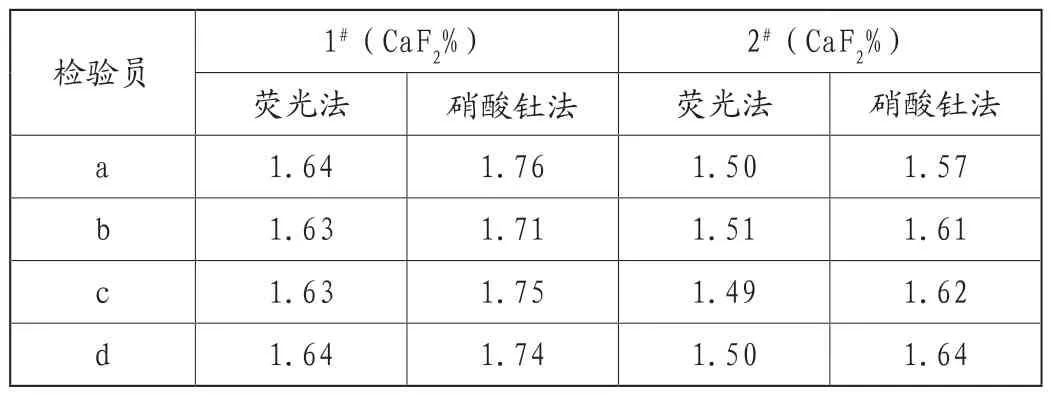
表2 样品中Cl-对结果的影响
(3)pH调节对结果的影响
按照硝酸钍的操作规程,溶液酸度范围在3.2~3.4之间。在2.2对样品处理的基础上,4个化验员对2个样品按照惯例采用广范试纸调节酸度并在同一实验室同一时间进行了分析,结果显示结果仍有较大差异。对4个化验员样品溶液的酸度利用采用pH计进行了准确测定,结果发现,4个化验员pH调节值范围差别较大,由此,可以判定采用广范试纸调节pH值导致结果出现了误差,为进一步确定酸度是影响硝酸钍滴定的关键因素,实验室对2个样品又采用pH计在不同酸度条件下由4个化验员分别进行了滴定,测得结果如表3所示。从实验结果可以看出,在相同pH条件下,由4个化验员采用硝酸钍滴定法获得的结果误差较小,而且与荧光法测定结果值很接近。同时发现,pH值越小,测定值越大,当用pH计准确控制酸度在3.2左右时测定结果误差较小,两种方法差异也较小。究其原因,广范pH试纸和pH计二者精密度本来就不同,前者并不能直观有效的调节到有效的pH值范围,再加上每个人的眼睛对颜色的分辨力本来也有一定的差别,导致结果异常也就成为常态化分析结果了[6]。因此,将广范试纸改为pH计,调节酸度在3.2左右,就会使分析结果更加准确可靠。
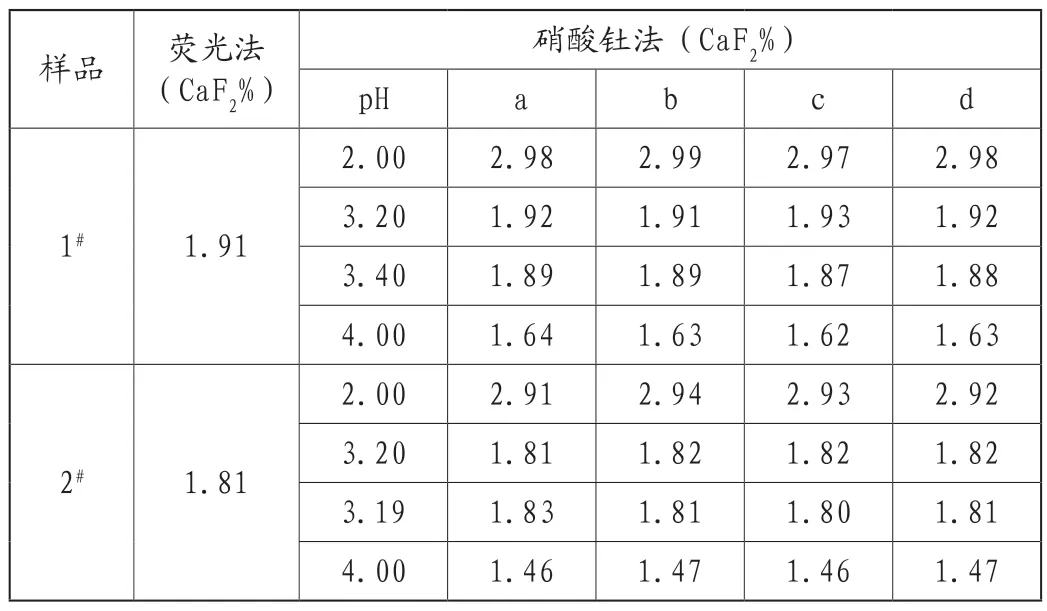
表3 pH调节对结果的影响
3.结论
为了进一步提高硝酸钍法测定钙含量的准确度,对硝酸钍法的操作步骤进行了进一步的优化,本试验以X射线荧光光谱法为对照,采用X射线荧光光谱法和硝酸钍法同时对样品进行测定。结果发现将一直沿用的采用广范pH试纸调节酸度改为pH计,并且准确调节pH到3.2左右,就能使硝酸钍滴定氟含量的分析结果与X射线荧光光谱法分析结果保持一致,准确度高。这一操作应该是影响硝酸钍法测定中钙含量的关键环节。但由于钍元素具有放射性,所以寻找其他物质替代硝酸钍应该是今后研究的方向。
猜你喜欢
云南化工(2021年11期)2022-01-12
食品安全导刊(2021年20期)2021-11-28
黄金(2020年11期)2020-09-10
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
食品安全导刊(2019年25期)2019-01-05
科学与财富(2018年14期)2018-06-11
凿岩机械气动工具(2017年1期)2017-05-17
中国塑料(2016年10期)2016-06-27
中成药(2016年8期)2016-05-17
河北地质(2016年2期)2016-03-20
