
I型神经纤维瘤病一家系基因检测
2024-02-26 13:06余佳琳梁嘉莉刘翌菲张辰希张晓颖李荣华
中国麻风皮肤病杂志 2024年3期
余佳琳 梁嘉莉 刘翌菲 蔡 妍 张辰希 张晓颖 刘 成 李荣华
1广东省东莞市大朗医院皮肤科,广东东莞,523770;2南方医科大学南方医院皮肤科,广东广州,510515;3广州医科大学附属武警广东省总队医院皮肤科, 广东广州, 510517;4福建医科大学附属泉州第一医院皮肤科,福建泉州,362000
I型神经纤维瘤病(neurofibromatosis type 1,NF1)是一种较常见的遗传性神经皮肤综合征,发病率为1/3500,主要表现为皮肤牛奶咖啡斑、腋窝雀斑、虹膜lisch结节及神经纤维瘤。本研究收集临床诊断NF1的患儿,经过二代测序及Sanger测序技术鉴定了NF1基因突变。现报道如下。
1 资料与方法
1.1 临床资料 患儿,女,10岁。因“全身多发咖啡斑10年,左眼睑及上颚肿物3年”就诊。患儿自出生起出现腋窝雀斑,牛奶咖啡斑,逐渐增多。3年前患儿出现左眼睑及上颚肿物,逐渐增长,无疼痛,无视力下降,无进食及吞咽困难。出生史无特殊,查体身高134.5 cm,体重28 kg,语言、运动及智力发育正常。皮肤科查体:全身散在皮肤咖啡牛奶斑,以躯干、腋窝、四肢等部位为主(图1a)。左眼上睑外侧睑板增厚,局部软组织增生,边界不清,表面组织无破溃,质韧、无压痛(图1b)。右侧上颌腭部一1 cm×0.8 cm粉色不规则肿物生长,质地柔软,边界不清,与周围组织粘连(图1c)。患儿父母正常,均无皮肤咖啡牛奶斑和肿物。检验检查:血常规、肝肾功能、凝血四项、术前四项及头颅MRI未见明显异常。右侧上颌牙龈肿物切除送病理活检,镜下见瘤细胞长梭形,细胞胞浆嗜伊红,细胞核长梭形,部分细胞核卵圆形,染色质细腻,未见核分裂象(图1d)。免疫组化示EMA(-)、Actin(SMA)(部分+)、S-100(+)、SOX-10(+)、CD34(+)、NP(局灶+)、Ki-67(+,约1%),符合神经纤维瘤。
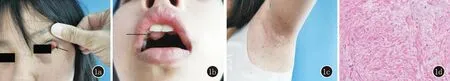
1a:左眼眶肿物;1b:上颚肿物;1c:腋窝雀斑;1d:组织病理学表现(HE,×200)
1.2 研究方法
1.2.1 基因组DNA 经患儿及家属知情同意,采集患儿及其父母外周静脉血,使用Qiagen DNA Mini Kit试剂盒(Qiagen,中国,上海)提取基因组DNA。
1.2.2 DNA文库制备 通过S220 Focused-ultrasonicator仪器(Covaris, Massachusetts, 美国)将1~3 μg的基因组DNA片段化至150 bp平均大小。使用文库制备试剂盒(MyGenotics,中国,北京),通过末端修复、接头连接和PCR扩增样本,制备DNA文库,通过DNBSEQ(DNBSEQ-T7)进一步测序。
1.2.3 目标基因捕获 使用骨骼疾病相关基因捕获试剂盒(MyGenostics Inc,中国,北京)对打断的DNA文库进行捕获。生物素标记的捕获探针设计包含所有基因的编码外显子以及外显子上下游50 bp侧翼区域,探针长度为100 bp。利用Illumina Nextseq 500第二代测序仪对捕获到的区域进行双端测序。
1.2.4 数据筛选与生物信息学分析 用cutadapt筛选MGI测序适配器和低质量读取(<80 bp)。在质量控制之后,使用Sention将读数映射到UCS hg19人类参考基因组。去除重复读取,使用参数驱动程序校正碱基,以便最终输出BAM文件读取中碱基的质量值更接近与参考基因组,使用映射读取来检测变异。最后通过Sentieon软件的参数驱动程序检测SNP和InDel的变异。将数据转换为VCF格式,使用ANNOVAR软件对变体进行了进一步注释(http://annovar.openbioinformatics.org/en/latest/),并与多个数据库关联,如千人基因组、ESP6500、dbSNP、EXAC、内部数据库(Mygenotics)、HGMD,并用REVEL、SIFT、PolyPhen-2、MutationTaster等进行变异有害性预测。
1.2.5 Sanger测序验证 获得所有可用家族成员的基因组DNA,根据需要测序的DNA片段合成引物,用聚合酶链反应(polymerase chain reaction, PCR)法进行扩增。将得到的产物进行纯化,纯化后的PCR产物经Sanger测序法(Biosune)测序。测序结果用DNASTAR(Madison)软件分析。
2 结果
2.1 测序数据分析 患儿基因检测平均测序深度为180.12,目标区域覆盖度为99.48%。
2.2 致病基因突变分析 全外显子测序显示患者17号染色体NF1基因内含子2发生c.204+2T>G位点杂合突变(图2a),导致第2个碱基由T变为G,为剪切突变,经过Sanger测序验证。根据ACMG指南,该变异初步判定为致病性变异(Pathogenic:PVS1+PS2+PS4+PM2_Supporting+PP4)。患儿母亲(图2b)和父亲(图2c)无该位点变异,患儿为自发杂合突变。

2a:先证者;2b:先证者的母亲;2c:先证者的父亲
3 讨论
NF1是一种常染色体显性遗传性肿瘤性疾病,50%为自发突变[2]。NF1以咖啡牛奶斑、神经纤维瘤和Lisch结节为特征,主要为皮肤、神经、眼、骨骼及血管等器官及系统受累的一种易患肿瘤综合症。
NF1临床症状主要为多发皮肤结节、咖啡牛奶斑、虹膜结节、腋下和腹股沟雀斑等特征性表现。根据美国国立卫生研究院制定及Legius更新的NF1诊断标准,满足以下2点即可诊断:(1)全身≥6处咖啡色斑,青春期前直径>5 mm,青春期后直径>15 mm;(2)≥2处任何类型的神经纤维瘤或1处丛状神经纤维瘤;(3)腋窝和(或)腹股沟雀斑;(4)视神经胶质瘤;(5)≥2个虹膜结节;(6)骨骼异常;(7)一级亲属患有NF1[3,4]。在本研究中,结合患儿临床体征及影像学检查资料,初步诊断为NF1。我们采用二代测序检测患者的全外显子,发现患者NF1基因内含子2发生杂合突变,c.204+2T>G杂合突变,导致氨基酸发生剪切突变。该患者父母未发生该位点突变,提示该突变为新发突变。
NF1基因定位于染色体17q11.2,跨越350 kb,包含60个外显子(其中3个为选择性剪接外显子),编码由2818个氨基酸组成的神经纤维蛋白[5]。神经纤维蛋白与RAS GTPase激活蛋白具有同源性,可通过加速Ras结合的GTP水解来负性调控RAS蛋白转导信号[6]。当NF1基因突变,神经纤维蛋白表达下调, Ras过度活化,下游通路持续激活,施万细胞过度增殖,形成神经纤维瘤[7]。NF1相关神经纤维瘤可发生于全身各个部位。例如本研究NF1患儿的神经纤维瘤发生于眼眶及上颚,影响患儿正常生活,首先手术治疗,上颚及眼眶中午经过手术治疗切除了肿物,但并不能预防复发。其他传统的治疗方法如激光治疗、放射疗法和化学疗法等,但效果有限且伴随着较大的创伤性[8,9]。这些方法无法阻止瘤体的新发[10],因此在临床上难以常规应用,需要探讨新的治疗方法。
NF1基因突变引起Ras/丝裂原活化蛋白激酶(MAPK)信号通路异常激活是目前较为认可的致病机制之一。因此,靶向抑制丝裂原活化蛋白激酶(MEK)是一种有效的靶向治疗方法。司美替尼是一种高度选择性口服小分子MEK抑制剂,能与MEK1/2的变构抑制结合位点特异性结合,阻止MAPK通路异常活化[11,12]。于2020年4月获得美国FDA批准,用于治疗2~18岁,有症状和/或进行性、不可手术的NF1相关神经纤维瘤[13]。还有许多MAPK处于研发及临床试验中,为NF1患者带来希望的曙光。
在本文中,我们对一例散发性的NF1患儿进行了基因检测,明确了致病基因,并鉴定出一个国内未曾报道过的新突变位点。这一发现丰富了NF1基因突变库,为NF1的遗传咨询和产前诊断提供了理论依据。另外,本研究患儿采用手术切除的方式去除了眼眶及上颚神经纤维瘤,此后仍需要门诊密切随访及多学科协助治疗,以便及时发现其他NF1临床特征,进而对疾病的严重程度做出评估,并较早采取合理的综合干预,提高患儿生存质量。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
今日农业(2022年15期)2022-09-20
农业工程学报(2022年12期)2022-09-09
中国生殖健康(2020年4期)2021-01-18
科教新报(2019年34期)2019-09-10
课堂内外(小学版)(2019年4期)2019-05-17
科学Fans(2019年2期)2019-04-11
中国生殖健康(2018年4期)2018-11-06
摄影之友(影像视觉)(2017年11期)2017-11-27
创新作文(小学版)(2017年24期)2017-02-25
