
组分百日咳抗原中间品中内毒素含量动态显色定量检测方法的建立及验证
2024-02-23 13:30:44赵明刘婷马昱吴丽洁付慧李世慧
中国生物制品学杂志 2024年2期
赵明,刘婷,马昱,吴丽洁,付慧,李世慧
北京生物制品研究所有限责任公司,北京 100176
百日咳是由百日咳杆菌引起的呼吸系统疾病,症状为长久的阵发性咳嗽,会导致严重的肺损伤,甚至危及生命,是儿童最常见的传染病之一[1-2]。我国在1995 年成功研制出无细胞百白破联合疫苗(diphtheria,tetanus and acellular pertussis combined vaccine,DTaP)[3],目前研制的吸附无细胞百(三组分)白破联合疫苗是DTaP 的更新换代,由百日咳类毒素(pertussis toxin,PT)、丝状血凝素(filamentous hemagglutinin,FHA)和百日咳黏附素(pertactin,PRN)抗原组成,经发酵、纯化[4]、脱毒、吸附后制备成原液,进而配制成疫苗。百日咳抗原中间品脱毒后,抗原在后续的原液吸附及半成品配制过程中不再经过去除内毒素的步骤,因此脱毒后抗原中细菌内毒素含量的高低直接影响最终疫苗成品中内毒素的含量。疫苗中内毒素含量较高时,注射进入人体内易引起发热、微循环障碍、内毒素休克及播散性血管内凝血等副反应[5-6],因此,建立一种灵敏度更高、能够定量检测组分百日咳抗原中间品中内毒素含量的方法,对于吸附无细胞百(三组分)白破联合疫苗生产过程中内毒素含量的质量控制至关重要。
《中国药典》三部(2020 版)<1143 细菌内毒素检查法>中的内毒素检测方法有凝胶法和光度测定法,凝胶法分为凝胶限度试验和凝胶半定量试验,光度测定法分为浊度法和显色基质法,显色基质法根据检测原理又分为终点显色法和动态显色法[7]。目前组分百日咳抗原中间品内毒素含量的检测方法为凝胶法之一的凝胶限度试验,通过将待测样品进行适宜倍数的稀释后进行检测,从而判断供试品的内毒素含量是否高于限值,该方法的弊端是不能准确测定出其具体含量,不利于进行中间品内毒素含量的过程控制以及成品吸附无细胞百(三组分)白破联合疫苗的质量控制。动态显色法是一种检测反应混合物的吸光度或透光率达到某一预先设定的检测值所需要反应时间或检测值增加速度的方法[7],已在食品、注射用水、血液等细菌内毒素检测的研究中广泛应用[8-12],但较少应用于生物制品中。该方法的优势在于其鲎试剂只需根据显色反应即可定量检测内毒素,准确度高、检测范围广,无需蛋白原凝固,抗干扰能力强,特别适用于生物样本的检测[13-15]。吸附无细胞百(三组分)白破联合疫苗的组分百日咳抗原中间品的内毒素检定尚未采用此方法。
本研究旨在建立动态显色法检测吸附无细胞百(三组分)白破联合疫苗的组分百日咳抗原中间品脱毒后内毒素含量,并进行验证,以期更好地对组分百白破疫苗进行质量控制。
1 材料与方法
1.1主要试剂及仪器 脱毒后的组分百日咳抗原中间品PT、FHA 和PRN 由北京生物制品研究所有限责任公司提供;Pyros Kinetix®Flex 96(PKF 96)细菌内毒素定量检测系统、动态显色法美洲鲎试剂(LAL,批号:2042103,灵敏度λ = 0.001 EU/mL)、葡聚糖抑制缓冲液(批号:1207080,规格:5 mL/瓶)、细菌内毒素工作标准品[(control standard endotoxin,CSE),批号:249035,规格:10 ng/瓶,10 EU/ng]、无热原水[(LAL reagent water,LRW),批号:AF29565799,规格:50 mL/瓶]、硼硅酸盐玻璃反应试管(8 mm×75 mm)和硼硅酸盐玻璃稀释试管(12 mm × 75 mm)均购自美国Associates of Cape Cod 公司。
1.2溶液的配制
1.2.1鲎试剂溶液 取葡聚糖抑制缓冲液3.2 mL,滴加至装有LAL 干粉的Pyrochrome®小瓶中,LAL 干粉在几分钟后进入溶解状态,完成复溶。在试验过程中按照样品体积0.2 mL,添加0.05 mL的鲎试剂。
1.2.2标准曲线溶液根据Cape Cod 公司的动态显色法细菌内毒素工作标准品标准化COA 文件制备50 EU/mL 的标准品贮备液:用2 mL 无热原水溶解1 瓶内毒素标准品(10 ng/瓶,10 EU/ng),充分振荡1 min,制成浓度为50 EU/mL 的标准品贮备液。用无热原水将50 EU/mL的内毒素标准品贮备液稀释成浓度为5、0.5、0.05、0.005 EU/mL 的标准品工作溶液。
1.2.3供试品溶液 将脱毒后PT、FHA、PRN分别添加无热原水,稀释不同倍数,PT:10、100、1 000倍;FHA:3 000、5 000、10 000倍;PRN:50、75、100倍。
1.2.4阳性对照溶液 取1.2.3 项制备的供试品溶液各0.9 mL,与0.1 mL 5 EU/mL 内毒素标准品溶液混合,作为供试品阳性对照。
1.3方法的建立分别取制备的标准曲线溶液、供试品溶液和阳性对照溶液200 µL 至硼硅酸盐玻璃反应试管中,以无热原水作为阴性对照。在Pyros®EQS v1.2检测软件上编辑检测序列,向每个反应试管滴加50 µL 鲎试剂溶液,振荡混合3 ~5 s,快速放入仪器内检测。待序列全部显示检测完成后,停止数据采集。软件自动计算供试品检测值、回收率[(阳性对照溶液内毒素浓度-供试品溶液内毒素浓度)/0.5×100%]和变异系数(CV)。
1.4方法的验证
根据《中国药典》三部(2020 版)<9101 分析方法验证指导原则>的相关要求,对百日咳抗原中间品中内毒素含量动态显色测定方法进行线性、专属性、准确性、精密性验证。试验采用0.005 ~5 EU/mL标准曲线计算供试品溶液内毒素含量。
1.4.1线性范围 对0.005 ~5 EU/mL 4个浓度的标准品工作溶液进行线性验证,以内毒素浓度为横坐标,反应时间的对数为纵坐标绘制标准曲线,得到标准曲线回归方程,结果应呈现为线性相关系数的绝对值(|r|)≥0.98,CV<20%,各浓度的标准品工作溶液回收率在80%~120%之间,阴性对照的反应时间小于0.005 EU/mL标准品溶液的反应时间。试验重复3次。
1.4.2专属性 先按下式确定最大有效稀释倍数(maximum valid dilute double,MVD)。
式中L 为脱毒后百日咳抗原中间品中细菌内毒素限值(检测浓度/蛋白质浓度= 500 EU/mg),λ为标准曲线最低浓度(0.005 EU/mL),c 为百日咳抗原中间品的蛋白质浓度(mg/mL)。
选择标准曲线中靠近中点的浓度(0.5 EU/mL)作为加标浓度(λm),取3 种不同稀释倍数的供试品0.9 mL,与5 EU/mL 内毒素标准品溶液0.1 mL 混合作为供试品阳性对照,无热原水为空白对照,进行干扰试验,计算回收率。
1.4.3准确性 确定脱毒后PT、FHA、PRN的无干扰浓度后,选择其中1个浓度用无热原水稀释(PT 100倍、FHA 10 000倍、PRN 50倍),各取0.9 mL,与5 EU/mL内毒素标准品溶液0.1 mL 混合作为供试品阳性对照,按确定的方法操作,每个样品重复检测6次,计算回收率(应在50%~200%之间),验证方法的准确性。
1.4.4精密性
1.4.4.1重复性 取脱毒后PT、FHA、PRN,分别用无热原水稀释100、10 000、50 倍后,各取0.9 mL,与5 EU/mL 内毒素标准品溶液0.1 mL 混合作为供试品阳性对照,按确定的方法操作,每个样品重复检测6次,计算CV(应<20%)。
1.4.4.2中间精密度 按上述方法制备供试品溶液及阳性对照,由不同操作人员于不同时间分别测定3次,计算CV。
1.5动态显色法定量检测与凝胶法检测结果的对比选取经凝胶法测定合格的脱毒后PT、FHA、PRN 各3批,分别用无热原水稀释100、10 000、50倍后,采用动态显色法检测内毒素含量,检测结果应小于脱毒后百日咳抗原中间品中细菌内毒素限值(500 EU/mg蛋白),且回收率应在50%~200%之间。
1.6数据采集及分析 脱毒后PT、FHA、PRN 稀释后作为供试品溶液,放入细菌内毒素定量检测系统中检测,使用Pyros®EQS v1.2 软件编辑序列,并进行数据采集及分析。
2 结果
2.1方法的验证
2.1.1线性范围 用无热原水稀释标准品溶液后,得到标准曲线的|r|均大于0.99,每个标准品浓度对应的回收率均在90%~115%之间,CV均小于6%,见表1。表明该方法在0.005 ~5 EU/mL 范围内线性良好。
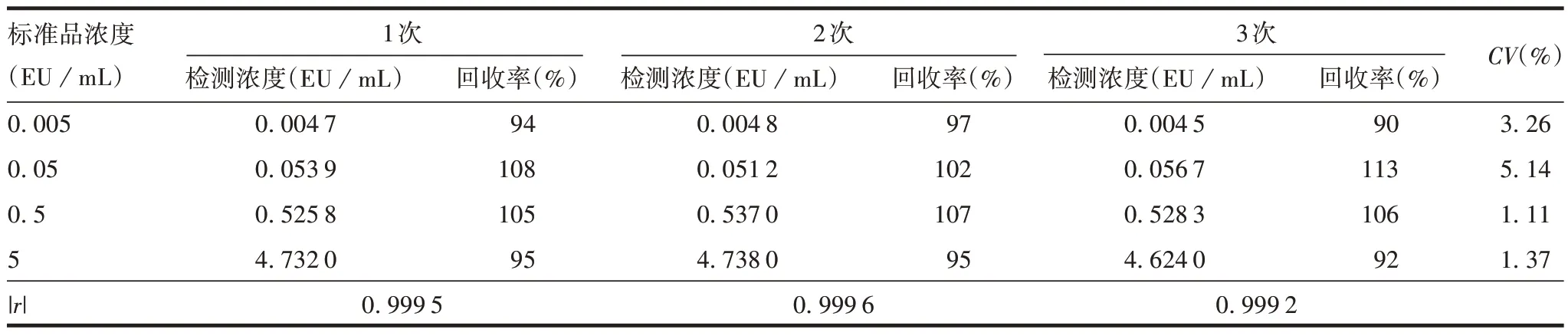
表1 线性范围验证结果Tab.1 Linear range verification results
2.1.2专属性 各供试品稀释对应的浓度回收率均在90%~120%之间,符合标准,见表2。表明百日咳抗原中间品脱毒后PT、FHA、PRN在对应稀释倍数下对细菌内毒素的含量检测无干扰,专属性良好。
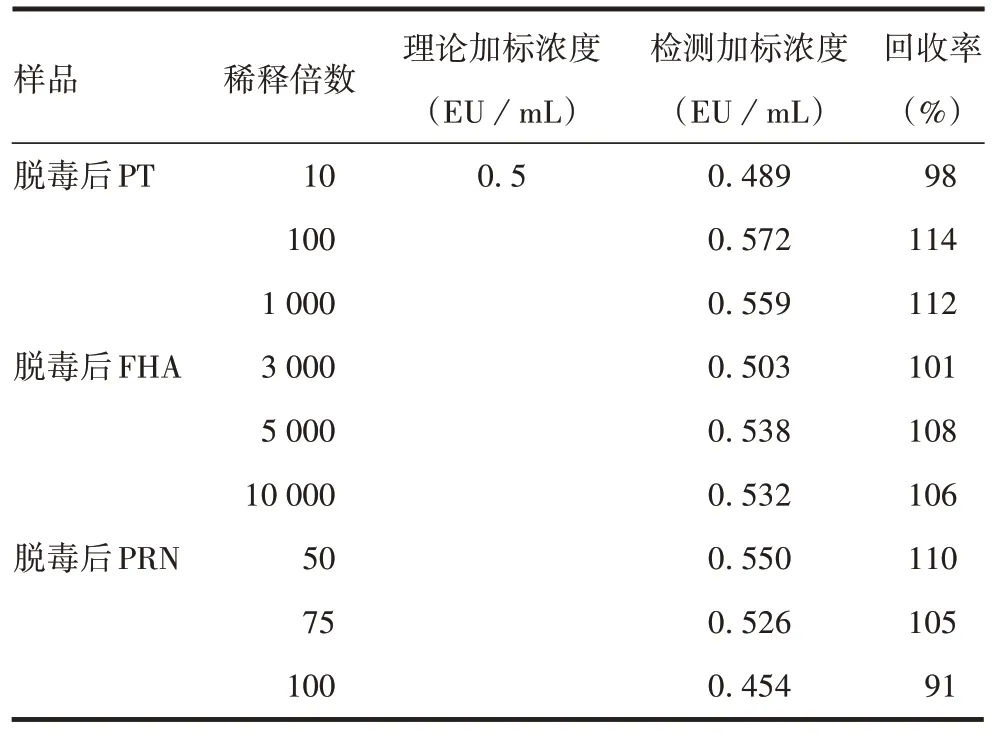
表2 干扰试验结果Tab.2 Interference test results
2.1.3准确性 脱毒后PT、FHA、PRN 测定结果的回收率分别为125%、110%、99%,均符合可接受标准,见表3。表明该方法准确性良好。
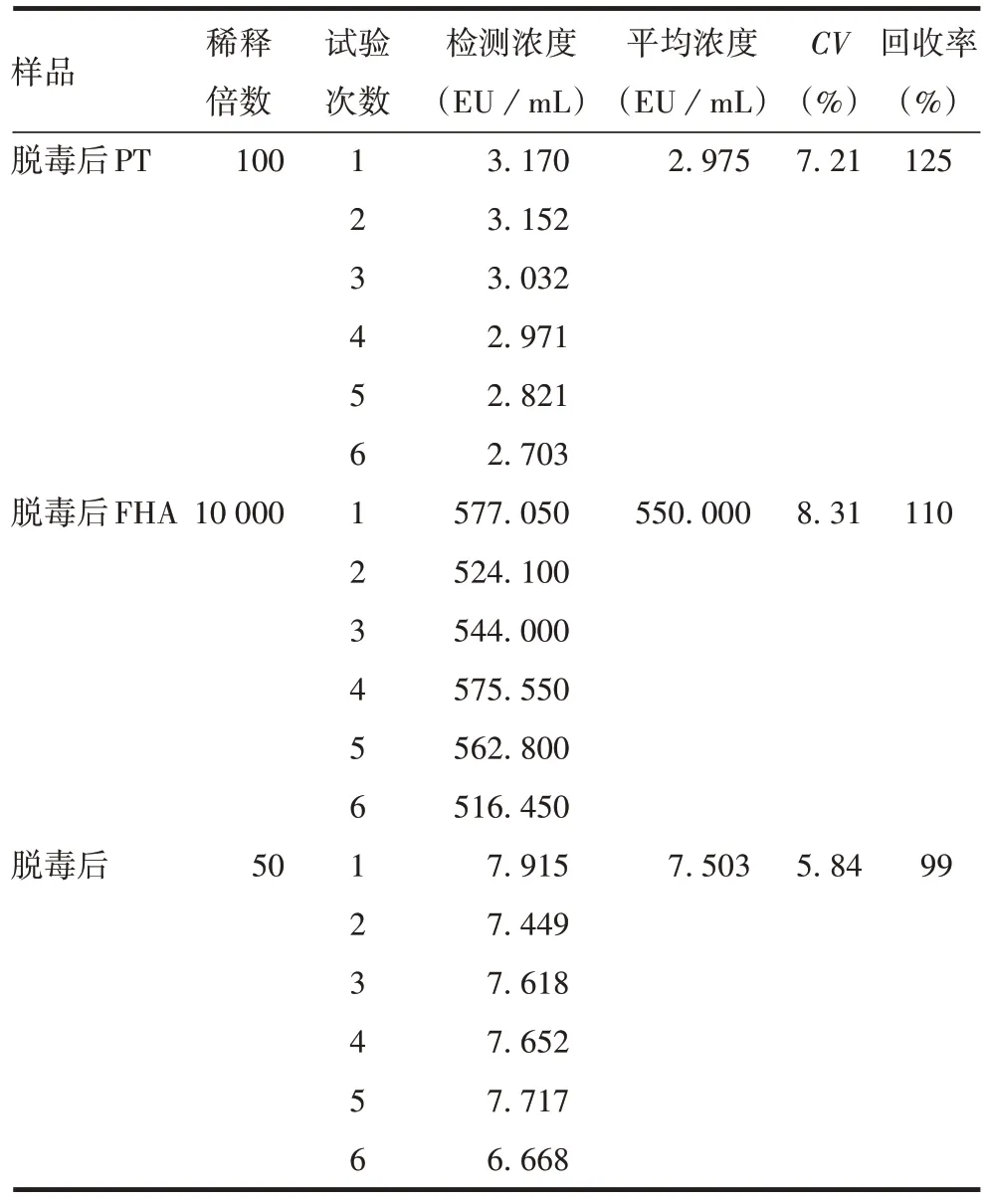
表3 准确性和重复性验证结果Tab.3 Accuracy and reproducibility verification results
2.1.4精密性 重复性验证脱毒后PT、FHA、PRN 测定结果的CV分别为7.21%、8.31%和5.84%,均小于20%,符合可接受标准,见表3,表明该方法重复性较好;中间精密度验证脱毒后PT、FHA、PRN 测定结果的CV分别为6.04%、16.29%和12.23%,符合可接受标准,见表4,表明该方法中间精密度较好。

表4 中间精密度验证结果Tab.4 Intermediate precision verification results
2.2动态显色法定量检测与凝胶法检测结果的对比采用动态显色法检测脱毒后PT、FHA、PRN供试品各3批的行内毒素含量,结果均小于脱毒后百日咳抗原中间品中细菌内毒素限值,且回收率在50%~200%之间,见表5,两种方法检测结果一致。
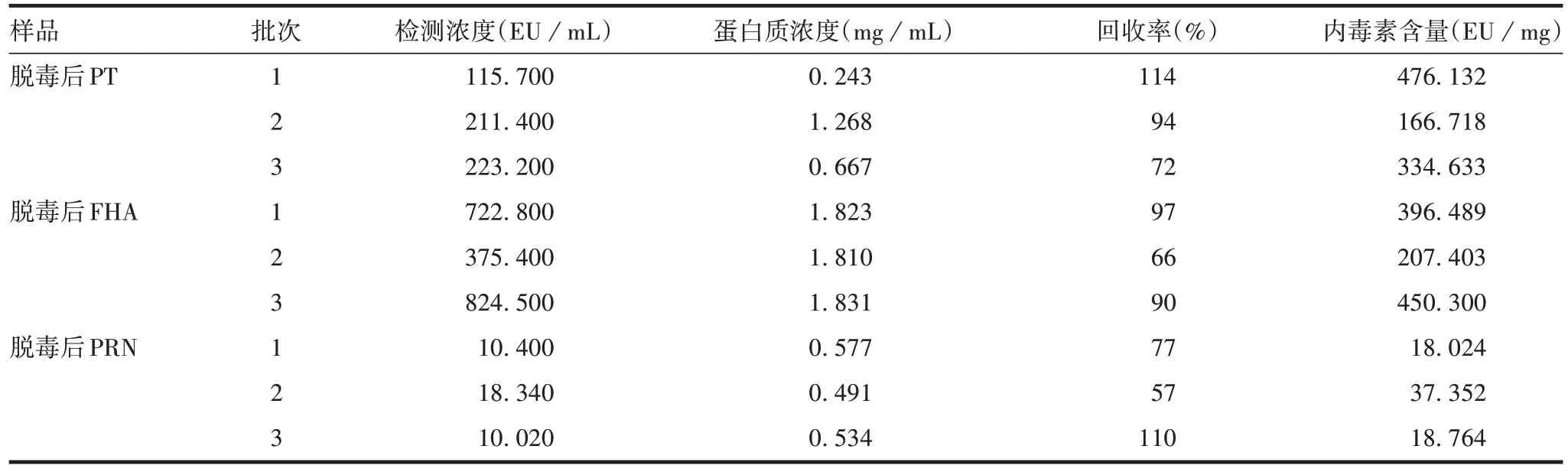
表5 动态显色法定量检测结果Tab.5 Quantitative detection results of kinetic chromogenic assay
3 讨论
细菌内毒素检查法已在一些领域中应用与研究[16-19],其中的光度测定法作为能够定量检测细菌内毒素含量的方法,已逐步应用于生物制品的内毒素检测研究[20-24]。光度测定法中的动态显色法具有能够定量、灵敏度高等优势,只需根据显色反应即能快速定量检测内毒素,灵敏度高达0.001 EU/mL,检测范围广,与鲎试剂配套的葡聚糖抑制缓冲液能有效消除干扰因素,操作简便[25]。因此,本研究建立了动态显色法检测百日咳抗原中间品的内毒素含量,并对该方法进行了验证,结果显示,在0.005 ~5 EU/mL范围内得到的标准曲线|r|均大于0.98,每个标准品浓度对应的回收率均在90% ~115%之间,CV均小于6%,表明该方法线性较好;脱毒后PT 稀释倍数为10、100、1 000,FHA稀释倍数为3 000、5 000、10 000,PRN稀释倍数为50、75、100,对细菌内毒素含量检测均无干扰作用;准确性和精密性验证结果均符合可接受标准。采用动态显色法对3 批脱毒后PT、FHA、PRN 进行细菌内毒素定量检测,其结果与凝胶法检测的同批供试品结果一致,证明该方法可行。
目前组分百日咳抗原中间品内毒素含量检测应用的是凝胶法,该方法不能准确定量,只能判断内毒素含量是否高于限值,不利于进行中间品内毒素含量的过程控制。本研究采用动态显色法检测组分百日咳抗原中间品脱毒后样品中的内毒素含量,能够进行定量,且方法具有良好的线性、专属性、精密性,检测灵敏、准确性高、范围广,与凝胶法相比,不仅能够判断结果是否合格,还能定量,且克服了凝胶法的主观判断因素,动态显色法也能应用于发酵、纯化、中间品及成品中百日咳抗原生产全过程的内毒素含量测定。建立的组分百日咳抗原中间品中内毒素含量定量检测方法,对组分百白破疫苗中间品生产的过程控制及成品的质量控制至关重要。
猜你喜欢
——基于进口关联化、多样化与高度化的多维视角
财经论丛(2023年5期)2023-05-08 13:28:28
厦门大学学报(哲学社会科学版)(2022年3期)2022-10-14 09:29:50
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:50
猪业科学(2021年3期)2021-05-21 02:06:18
心肺血管病杂志(2020年5期)2021-01-14 00:43:52
山东社会科学(2020年1期)2020-12-20 18:51:22
健康博览(2019年3期)2019-12-02 05:42:16
大众医学(2019年11期)2019-01-03 06:56:43
现代商贸工业(2017年24期)2017-09-12 21:19:38
中国神经免疫学和神经病学杂志(2017年3期)2017-01-12 12:44:57
