
小汗腺汗孔癌三例
2024-01-30 08:33蒋佳红高丽娜顾安康郭海霞
中国麻风皮肤病杂志 2024年2期
蒋佳红 高丽娜 顾安康 张 俊 郭海霞
1天津中医药大学研究生院,天津,301617;2天津市中医药研究院附属医院,天津,300120
小汗腺汗孔癌是一种起源于汗腺表皮内导管上皮的恶性皮肤附属器肿瘤,临床罕见,临床表现缺乏特异性,现报道3例临床表现不同的小汗腺汗孔癌。
临床资料患者1,女,46岁。腰背部棕红色结节2年。2年前患者无明显诱因于腰背部右侧发现一黄豆大小孤立结节,无明显自觉症状,未诊治,后皮损渐渐增大、隆起,偶有压痛,遂就诊于我科。患者既往体健。
皮肤科检查:腰背部右侧一约1.8 cm×1.7 cm大小红褐色斑块,表面可见棕褐色点,粗糙呈疣状,边界清,无糜烂、破溃(图1a)。组织病理示:表皮变化不明显,真皮内可见大小不等的瘤细胞团呈巢状分布。瘤细胞似汗孔样细胞,体积较小,排列紧密,可见空泡化透明细胞和导管分化,瘤巢内细胞异型性明显,胞核大而深染、形状不规则,呈侵袭性生长,瘤团中偶见片状坏死(图1 b、1c)
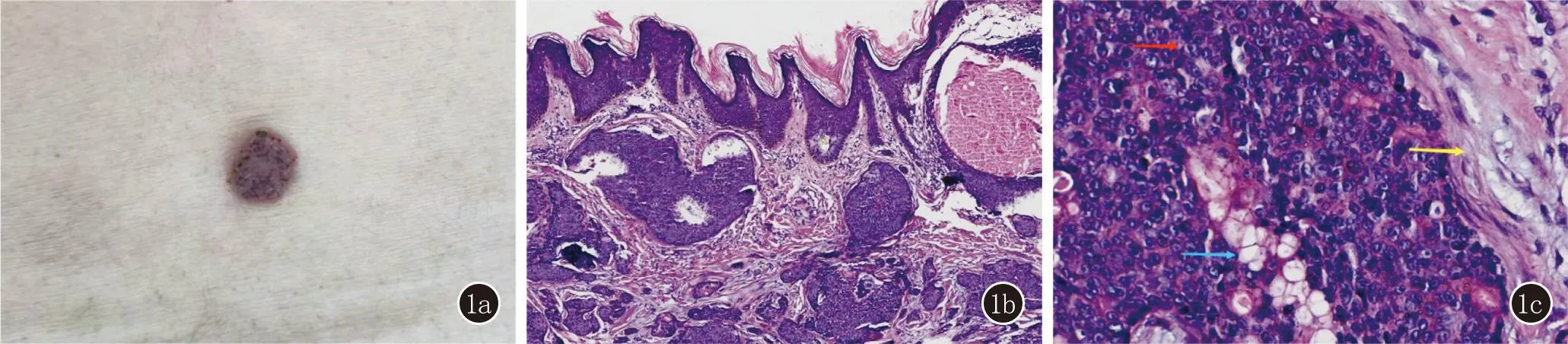
图1 1a:腰背部右侧红褐色斑块;1b:表皮变化不明显,真皮内可见大小不等的瘤细胞团呈巢状分布(HE,×20);1c:瘤细胞似汗孔样细胞,细胞异型性明显(红箭头),空泡化透明细胞(蓝箭头),间质组织条索状浸润(黄箭头)(HE,×200)
患者2,男,80岁。右手掌部息肉样增生物10余年,迅速增大破溃2个月。患者10余年前无明显诱因右手掌心出现一粟粒大小红色丘疹,无明显不适,未予重视,后皮损缓慢增大,并渐呈息肉样外观。就诊前2个月,无明显诱因,皮损迅速增大至蚕豆大小,伴表面破溃,少许渗出。当地医院抗感染等局部对症治疗无明显改善,遂来我院就诊。既往高血压病史20余年,规律服用降压药,血压控制良好,否认其余病史。
皮肤科检查:右手掌心一粉色半球状息肉样增生物,大小约1.5 cm×1.0 cm,表面破溃,可见少许清亮及血性渗出液,增生物根部与基底连接紧密,基底周围皮肤角化隆起呈堤状(图2a)。

图2 2a:右手掌心粉色半球状息肉样增生物;2b:表皮增厚,与表皮相连的肿瘤细胞条索状向真皮内浸润性生长(HE,×20);2c:内衬嗜酸性护膜的导管样结构(红箭头)(HE,×200)
组织病理示:表皮增厚,可见与表皮相连的肿瘤细胞条索状向真皮内浸润性生长,肿瘤细胞形态似汗孔样细胞,体积较棘细胞小,排列紧密,与表皮界限清晰,部分瘤细胞异型性明显,胞核大而深染、形状不规则,部分瘤团中可见片状细胞坏死、空泡化透明细胞和内衬嗜酸性护膜的导管样结构,间质疏松水肿(图2b、2c)。
患者3,女,68岁,右大腿斑块3年。患者3年前无明显诱因于右大腿屈侧发现黄豆大小褐色类圆形肿物,后皮损渐增大,偶伴瘙痒及疼痛,遂就诊于我科。患者幼时因疾病发热后聋哑。
皮肤科情况:右大腿屈侧褐色肿物,大小约5.0 cm×3.5 cm,皮损隆起,表面破溃、出血,边界清(图3a、3b)。

图3 3a、3b:右大腿屈侧褐色类圆形肿物;3c:表皮增厚,表皮及真皮内大小不等、界限清晰的瘤团(HE,×40);3d:瘤巨细胞形成(红箭头),细胞角化(黄箭头),空泡化细胞(蓝箭头)(HE,×400);3e:CK5/6(+)(免疫组化,×100);3f:P63(+)(免疫组化,×100);3g:Ki-67 index约30%(免疫组化,×100)
组织病理示:表皮增厚,表皮及真皮内可见大小不等的瘤团,正常表皮与肿瘤团块之间界限清晰,瘤巢周边细胞形态似汗孔样细胞,中央细胞核大、深染,病理性核分裂象多见,散在瘤巨细胞,瘤巢内可见充分发育的导管结构及正在形成导管的空泡化细胞。免疫组化:CK5/6(+),P40(+),P63(+),Ki-67 index约30%,P53 index约10%,PAS(-),EMA(-),CEA(-)(图3c~3g)。
3例患者家族中均无类似病史,体格检查生命体征平稳,各系统检查无异常。结合临床表现及组织病理3例患者均诊断为小汗腺汗孔癌,行局部皮损扩大切除术,例3联合皮下蒂皮瓣修复创面(图3b),随访半年至两年均未复发。
讨论小汗腺汗孔癌(eccrine porocarcinoma,EPC)是一种起源于汗腺表皮内导管上皮的恶性皮肤附属器官肿瘤,临床罕见,好发于头颈部(39%)和下肢(29%),其次为躯干(17%)[1]。发病率约为(0.02~0.2)/105/年,仅占所有皮肤恶性肿瘤的0.005%~0.01%,性别间发病率无明显差异[2]。本病多见于60岁以上的老年人,年轻患者少有报道,由小汗腺汗孔瘤(eccrine poroma,EP)进展为EPC的8岁儿童是目前已报道的年龄最小患者[3]。
EPC的病因与发病机制尚不明确,慢性紫外线暴露、免疫抑制是EPC发生的危险因素。现有研究发现表皮生长因子受体、肿瘤蛋白p53在EPC中呈高表达,细胞周期蛋白依赖性激酶抑制剂2A以及丝裂原激活蛋白激酶通路参与EPC的发生机制[4]。本病可起始即为恶性,或由EP、脂溢性角化病、皮脂腺痣等良性肿物转化[5,6]。EP恶性转化平均时间约8.5年,原发良性皮损短时间内快速生长、瘙痒、自发溃疡或多结节出现提示恶变的风险,在鉴别诊断中需考虑,如患者2。
EPC缺乏特异性临床表现,可表现为疣状红斑、结节或息肉状皮损,伴或不伴溃疡和自发性出血及不同程度的瘙痒、疼痛,需结合组织病理学检查进行诊断。其病理诊断依据主要为:①与汗孔瘤相似的肿瘤细胞形态和增生模式;②瘤细胞侵袭性生长,异型性明显, 胞核大、深染,形状不规则,可形成瘤巨细胞;③瘤团内汗腺分化的导管样结构。此外,病理改变还包括瘤团局限于表皮或扩展至真皮,可见片状瘤细胞坏死、广泛性透明细胞改变,或伴有间质结缔组织增生。免疫组化无特异性,可出现上皮膜抗原(EMA)及癌胚抗原(CEA)表达阳性。
目前国内外学者对EPC的辅助检查有了进一步的认识。头部EPC超声表现为低回声团,边界清晰,内回声不均,可见点状强回声,彩色多普勒血流成像于肿块内部及周边探及较丰富血流信号[7]。皮肤镜检查可见多形血管,包括发夹状、分枝状和点状血管,以及呈圆形-椭圆形粉红色-白色结构区和白色-粉色晕圈组合结构,可用于EPC的诊断,但其准确性有待证实。
EPC为低度恶性肿瘤,及早行局部扩大切除术或莫氏显微外科手术是一线疗法。对于发生在皮肤张力较小的大面积皮损,可选择适宜的皮瓣,如患者3采用皮下组织蒂皮瓣。本病约有20%出现复发或转移,最常见的是区域淋巴结,远处转移包括肺、肝和脑转移。放疗和化疗对于预防复发和转移效果不佳,但在转移性EPC病例中,针对EGFR/MAPK通路的靶向治疗效果明显[4],需大规模临床研究进一步验证其有效性和安全性。
猜你喜欢
中国人民公安大学学报(自然科学版)(2022年3期)2022-10-18
刑事技术(2022年3期)2022-06-10
皮肤病与性病(2021年3期)2021-07-30
动漫界·幼教365(小班)(2021年11期)2021-03-23
传感器与微系统(2018年5期)2018-04-27
中国司法鉴定(2016年4期)2016-08-23
小朋友·快乐手工(2016年9期)2016-05-14
中华皮肤科杂志(2014年3期)2014-12-19
西南军医(2014年4期)2014-01-19
数学大世界·小学低年级辅导版(2009年8期)2009-07-28
