
混合谱系激酶结构域样蛋白非坏死性凋亡功能的研究进展*
2023-12-04 02:10:18沈钺陈东良陆远富李夏
中国病理生理杂志 2023年11期
沈钺, 陈东良, 陆远富, 李夏
(遵义医科大学基础药理教育部重点实验室暨特色民族药教育部国际合作联合实验室,贵州 遵义 563000)
坏死性凋亡(necroptosis)是Degterev等[1]于2005年命名的一种细胞程序性死亡形式。细胞凋亡受阻时,坏死性凋亡作为一种备用途径确保细胞死亡的进展[2]。不同于凋亡对胱天蛋白酶(caspase)活性的依赖,坏死性凋亡的发生依赖于混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein,MLKL)的磷酸化。MLKL属于蛋白激酶超家族,是一种不具有激酶功能的假激酶,作为终末专属效应蛋白在坏死过程中发挥作用[3-4]。坏死性凋亡启动后,MLKL与受体相互作用蛋白激酶1(receptor-interacting protein kinase 1, RIPK1)和RIPK3共同形成坏死体,并被RIPK3磷酸化,磷酸化的MLKL同源寡聚后易位至质膜并破坏其完整性以诱导细胞坏死性凋亡[5]。与非免疫原性的凋亡不同,坏死性凋亡可导致细胞内成分外泄从而引起强烈的炎症反应[1,6],参与缺血性再灌注损伤[7]、肾小管间质纤维化[8]和肝脏疾病[9]等多种炎症性疾病的发生发展。
MLKL是唯一可以有效执行坏死性凋亡的分子[3-4],一度被视作坏死性凋亡相关疾病的治疗靶标。但越来越多的研究证实MLKL在非坏死性凋亡事件中同样发挥重要作用,以MLKL为靶点抑制坏死性凋亡的同时也可扰乱其介导的其他功能。因此,本综述将从MLKL的坏死性凋亡相关功能出发,梳理MLKL在自噬、内体转运等非坏死性凋亡功能方面的研究进展,探析MLKL发挥不同功能的机制,为MLKL的深入研究及相关疾病干预策略的合理制定提供启示。
1 MLKL的坏死性凋亡相关功能
MLKL结构特点决定了其介导的坏死性凋亡激酶级联反应的发生。N末端的四螺旋束(four-helix bundle, 4HB)和C末端的假激酶结构域(pseudokinase domain, PsKD)为MLKL的功能结构域,两者由两个α螺旋组成的支撑结构域连接[10]。4HB结构中的疏水部分可与质膜的磷脂双分子层发生相互作用,实现与质膜的结合,此疏水特性是决定MLKL膜定位及膜结合能力的关键[11-12]。因此,4HB也被称为坏死性凋亡的“刽子手”结构域。PsKD在调控MLKL释放4HB中发挥分子开关作用,即调控MLKL在活化构象和非活化构象之间的切换[10]。支撑螺旋则充当信号传递器,将PsKD的磷酸化事件传递给4HB结构域[13],最终推动4HB结构域与质膜结合,从而介导坏死性凋亡的发生[14]。
MLKL磷酸化介导坏死性凋亡发生的分子机制依赖于RIPK3。RIPK3可磷酸化小鼠MLKL的第345位丝氨酸(Ser345)、Ser347和第349位苏氨酸(Thr349)[5]或人类MLKL的Ser357和Thr358[12],诱导PsKD构象发生变化并解除4HB结构域的自抑制[15-16]。活化后的MLKL在受体酪氨酸激酶TAM(Tyro3, Axl and Mer)的进一步作用下发生寡聚,寡聚体向质膜转移[11,17]。MLKL的4HB结构域中带正电荷的残基区域嵌入质膜带负电荷的磷脂酰肌醇磷酸盐或心磷脂[18],形成孔道[19]。这些孔道允许胞外离子内流导致细胞肿胀和膜溶解,最终造成细胞坏死性凋亡[20]。
2 MLKL的非坏死性凋亡相关功能
随着研究的深入,MLKL的非坏死凋亡相关功能也逐渐在多种病理生理事件中显现。MLKL的非坏死性凋亡作用与坏死性凋亡并无直接关系,两者可伴随也可不伴随发生。
2.1 MLKL磷酸化调节自噬 自噬是细胞利用溶酶体降解和回收内部成分的机制[21]。坏死性凋亡过程中常伴有自噬通量的改变。Ogasawara等[22]的研究显示,RIPK3可与自噬体标志蛋白——微管相关蛋白1轻链3-II(microtubule-associated protein 1 light chain 3-II, LC3-II)竞争SQSTM1/p62蛋白复合物(简称P62),形成RIPK3-p62复合体。此种结合隔离了LC3-II与p62的相互作用,阻碍自噬体在溶酶体中降解,最终造成自噬通量的抑制[22]。也有研究表明,触发RIPK1/RIPK3依赖性坏死性凋亡伴有自噬增强,而这一自噬促进作用与细胞外信号调节激酶(extracellular signal-regulated kinase, ERK)的激活有关[23-24]。此外,MLKL可独立于坏死性凋亡调节自噬。Wu等[25]观察到,在棕榈酸(palmitic acid, PA)刺激下,MLKL磷酸化后与自噬体发生共定位,进而导致自噬体与溶酶体的融合受到阻碍,抑制自噬。并且,该研究还证实了自噬抑制剂氯喹和亮肽素对自噬的抑制作用依赖于MLKL的表达,Mlkl的敲除可导致这两种自噬抑制剂的作用无法发挥。然而,也有研究指出,MLKL磷酸化能促进转运必需内体分选复合物(endosomal sorting complex required for transport, ESCRT)介导的膜切割过程中自噬体内外膜的分离,使自噬体与溶酶体的融合和随后的物质降解增强,从而增加自噬通量[26]。上述研究中MLKL在调节自噬方面表现出的双向调控作用可能与实验细胞类型及刺激手段的不同有关,但具体的调控机制仍需进一步研究。
2.2 MLKL磷酸化调节内体转运 哺乳动物细胞内存在着复杂多样的内吞途径,内体转运在细胞生物合成途径中发挥着重要的调控作用[27]。研究显示,在细胞稳态条件下MLKL可独立于RIPK3活性而参与内体运输过程的调节。磷酸化的MLKL能进一步增强含有磷酸化MLKL的细胞外囊泡的产生与释放,从而降低胞内磷酸化MLKL水平,限制其介导的质膜渗透和坏死性凋亡的发生[28]。Gong等[29]的研究表明,MLKL利用囊泡向胞外释放磷酸化MLKL的能力与ESCRT-III有关。ESCRT-III作为MLKL磷酸化后的下游途径,介导了含有受损质膜的“气泡”的形成,这一过程有助于维持质膜完整性以延缓细胞死亡。MLKL磷酸化后促使囊泡向胞外释放磷酸化MLKL的机制为细胞产生相对更多的炎症细胞因子和趋化因子创造了额外的时间窗口,有利于机体对坏死细胞的识别和清理[29-30]。
2.3 MLKL核易位调节细胞死亡和基因表达 除易位到质膜,MLKL磷酸化后还能易位至细胞核发挥不同的调控功能[31]。研究表明,在坏死性凋亡期间,RIPK3和MLKL在细胞核和细胞质之间持续穿梭。当核输出受到抑制时,RIPK3和MLKL在细胞核中大量积累,在此过程中,胞质中MLKL寡聚体组装受阻,减少了细胞的坏死性凋亡[32]。该研究提示坏死性凋亡信号成分的核易位是调节细胞坏死性凋亡的机制之一。MLKL也被证实能以先天免疫传感器ZDNA结合蛋白1(Z-DNA binding protein 1, ZBP1)/RIPK3依赖的方式活化并发生核易位,核内MLKL破坏核膜触发一种由核内到核外的死亡途径[33]。此外,Dai等[34]指出,MLKL可与RNA结合基序蛋白6(RNA binding motif protein 6, RBM6)相互作用而形成MLKL-RBM6复合物,该复合物入核后通过增加mRNA稳定性促进细胞间黏附分子1(intercellular adhesion molecule-1, ICAM-1)、血管细胞黏附分子1(vascular cell adhesion molecule-1, VCAM-1)和E-选择素在内皮细胞中的表达。总之,MLKL在细胞核的功能不仅涉及调节细胞死亡机制,还能影响细胞基因的表达。
2.4 MLKL线粒体易位调节活性氧(reactive oxygen species, ROS)生成 线粒体ROS增加是细胞发生坏死性凋亡时伴有的常见现象,这被证实与MLKL的线粒体易位有关[35-36]。Srivastava等[37]的研究证明,慢性肾病中的持续炎症会诱导RIPK3-MLKL复合物易位至线粒体,导致ROS增多并激活丝氨酸-苏氨酸激酶和钙/钙调蛋白依赖性蛋白激酶II,促进α-平滑肌肌动蛋白、胶原蛋白等的表达,加速疾病进展。也有研究显示,抑制MLKL向线粒体易位有助于恢复酒精所致的肝细胞线粒体功能障碍[38]。敲减Mlkl可降低细胞内ROS水平并在一定程度上减少ROS诱导的细胞死亡[39-40]。以上研究结果表明,MLKL的线粒体易位可能是坏死性凋亡期间线粒体功能障碍的关键诱发者。但是,在一项有关MLKL在人类血小板中作用的研究中,Ekhlak等[41]观察到抑制MLKL不仅显著延缓凝血酶诱导的血小板止血反应,还造成血小板线粒体跨膜电位被破坏、质子泄漏增加和ROS升高,MLKL展现出了稳定血小板线粒体功能的作用。上述研究提示,不同研究条件下MLKL对线粒体功能的影响不同,需根据不同的实验条件进一步探讨其对线粒体具体功能的影响。
2.5 MLKL影响巨噬细胞功能 巨噬细胞是传统的先天免疫细胞,在清除病原体和维持组织稳态方面起着至关重要的作用[42-43]。MLKL对巨噬细胞功能具有显著影响,当Mlkl基因敲除或MLKL活性受到抑制时,髓系巨噬细胞吞噬大肠杆菌生物颗粒的能力会被削弱[44],由此表明MLKL在维持巨噬细胞的吞噬功能中发挥重要作用。并且,在与巨噬细胞脂质代谢相关的机制中,敲减Mlkl会导致动脉粥样硬化斑块内及外周巨噬细胞脂质运输异常,造成巨噬细胞中脂质蓄积以及向泡沫细胞转变的增加[45]。另外,MLKL也参与巨噬细胞的表型转化。Mlkl缺失可推动缺血皮层中小胶质细胞从M1型向M2型的转化,进而起到神经保护作用[46]。这些研究结果揭示了MLKL在巨噬细胞中的多重积极作用。
3 MLKL发挥非坏死性凋亡功能的潜在机制
3.1 磷酸化位点 MLKL具有多个磷酸化位点,而每个位点的磷酸化都可赋予它独特的生物学效应。表1中总结了目前已鉴定的MLKL磷酸化位点及其相应的功能特性。
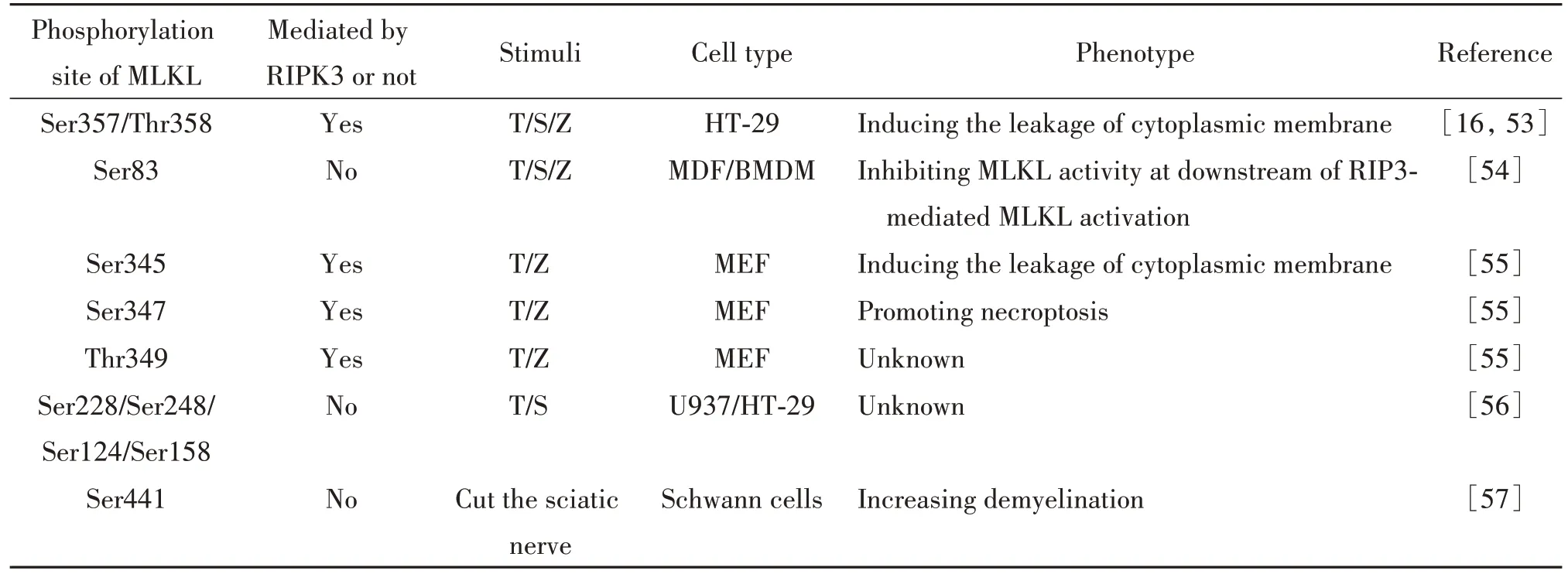
表1 MLKL不同磷酸化位点所表现出的作用Table 1. The roles exhibited by different phosphorylation sites on MLKL
多项研究显示,在不同的细胞类型中,MLKL磷酸化后常伴随着易位的发生[11,16,47-48]。蛋白磷酸化修饰后的易位主要是通过调节其电荷特性[49-50]、构象或结构性质[51-52]等来实现的。已有数据支持MLKL的易位与其磷酸化状态有关。据报道,MLKL抑制剂GW806742X能够以ATP依赖性方式与MLKL的PsKD结合,进而抑制MLKL磷酸化,阻止其向质膜转移[11],这强化了MLKL易位的磷酸化依赖性特征。因此,一方面,MLKL的功能与其特定的磷酸化位点有关;另一方面,多个磷酸化位点的存在或许是MLKL易位驱动力的重要来源。
3.2 亚细胞定位 MLKL的4HB结构域主要负责与膜的连接,其活化后可与易位途中碰到的细胞器膜结合[12,47]。细胞坏死性凋亡过程中,MLKL可易位至多个亚细胞结构,如内质网、线粒体[16]、外泌体[28,58]、细胞核[31-32]和内体[28-29,59]等。即便在非坏死性凋亡情境下,研究者也能观察到MLKL与多个亚细胞结构的共定位[25,44]。
MLKL的功能多样性与其在各亚细胞结构中的共定位关系紧密。当MLKL活化并易位至内质网膜时,可通过不干扰未折叠蛋白应答(unfolded protein response, UPR)信号传感器与葡萄糖调节蛋白78(glucose-regulated protein 78, GRP78)结合的方式激活内质网膜应激信号以调节蛋白质折叠的功能,这与常规的内质网应激反应需要GRP78从UPR传感器中解离不同[60]。同样,感染乙型肝炎病毒和黄曲霉毒素会上调环加氧酶2(cyclooxygenase-2, COX-2),导致RIPK3和发动蛋白相关蛋白1(dynamin-related protein 1, Drp1)表达增加,促使RIPK3-MLKL易位至线粒体并损害其功能,从而引发脂质代谢紊乱[61]。在上述研究中,MLKL对内质网和线粒体功能的影响与其易位后破坏细胞器膜有关。然而,关于MLKL在溶酶体定位后的效应,目前观点尚未达成统一。有研究指出,MLKL磷酸化后会易位至溶酶体并增加溶酶体膜通透性,致使溶酶体功能紊乱[62]。也有研究证实活化的MLKL与溶酶体共定位后不会对溶酶体的pH和数量产生明显影响[16,63],因此,仍需进一步研究确认MLKL影响溶酶体功能的机制。此外,MLKL与蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase, PERK)/真核翻译起始因子2α(eukaryotic translation initiation factor 2α, eIF2α)通路之间存在相互调控。MLKL可促进PERK/eIF2α通路的激活,而该通路的激活又可加强MLKL的核易位并以一种与核膜破坏无关的方式促进细胞凋亡[64]。由此可见,特定条件下向不同细胞器的易位可能是MLKL功能多样化的关键因素之一。
3.3 MLKL的非经典激活途径 MLKL的经典活化途径依赖于RIPK3。2012年,Sun等[53]首次明确了MLKL可以被RIPK3招募并磷酸化以执行坏死性凋亡功能,同时也提出了“MLKL可不依赖于RIPK3调控信号转导,因为它在不同细胞类型中比RIPK3更广泛地表达”的设想。随着MLKL非依赖RIPK3的激活方式的确认,这一设想逐步得到了证实。
Günther等[9]的研究证实,信号转导及转录激活因子1(signal transducer and activator of transcription 1, STAT1)的激活可增加MLKL的蛋白水平。因此,STAT1被视作MLKL的一个重要调节因子。同时,Wu等[44]也指出,抑制STAT1可下调MLKL的磷酸化水平,而单独抑制RIPK3仅能部分削弱MLKL的磷酸化,表明MLKL至少存在RIPK3依赖的和STAT1依赖的两种磷酸化途径。另外,Zhan等[26]的研究显示,在表达RIPK3的小鼠成纤维细胞L929和不表达RIPK3的小鼠神经母细胞瘤N2a细胞中,短期饥饿处理均可导致MLKL以依赖于钙/钙蛋白依赖性蛋白激酶II的方式发生磷酸化。以此种方式磷酸化的MLKL不仅不会诱导细胞的坏死性凋亡,反而能通过促进自噬体成熟来增加自噬通量,最终减缓细胞死亡。此外,磷脂酰肌醇3-激酶(phosphatidylinositiol 3-kinase, PI3K)/蛋白激酶B(protein kinase B, PKB/AKT)也被报道能直接触发MLKL的磷酸化并促进血小板向活性止血单位的转化[41]。该研究显示,抑制PI3K/AKT可明显减少凝血酶诱导的MLKL磷酸化,而抑制RIPK3则对MLKL的磷酸化水平影响甚微,揭示了一种与RIPK3无关但依赖PI3K/AKT的MLKL磷酸化途径。
除上述研究外,一项有关坐骨神经损伤的研究也报道了MLKL的非RIPK3磷酸化机制。研究者观察到MLKL在Schwann细胞轴突损伤后被诱导,并通过Ser441位点磷酸化;激活后的MLKL易位至髓鞘膜与髓磷脂结合,以破坏膜结构的方式加速髓磷脂分解,促进神经再生[57]。该研究证实MLKL促进神经再生的作用并不依赖于RIPK3,但这一过程中诱导MLKL磷酸化的激酶尚未可知。
MLKL存在多个非依赖于RIPK3的激活途径。当MLKL通过这些非经典激活途径激活时,其所展现出的功能与坏死性凋亡过程中导致细胞破裂死亡的结局截然不同,这可能是MLKL多样化功能产生的另一个关键因素。
4 总结
如上所述,MLKL的非坏死性凋亡相关功能复杂多样,具有多个磷酸化位点、能够与不同的亚细胞器发生共定位以及非依赖RIPK3的激活途径的存在可能是MLKL产生不同功能的关键因素。目前,MLKL的非坏死性凋亡相关功能虽然被广泛研究报道,但这些研究大多集中在表达水平及功能,对分子机制的研究仍不够深入。因此,未来无论是在坏死性凋亡还是非坏死性凋亡的相关功能研究中,都应注意明确MLKL的磷酸化位点及磷酸化后可能带来的功能转型;同时,MLKL在细胞内的亚定位以及与其他分子或亚细胞结构共定位后产生的潜在的生物效应也应被作为研究的重点;此外,还需深入挖掘除RIPK3以外的MLKL磷酸化激酶。总之,MLKL的研究尚需广泛探索,这是将MLKL作为疾病治疗靶点的前提。
猜你喜欢
中国卫生标准管理(2022年21期)2023-01-03 02:36:34
现代畜牧科技(2021年9期)2021-10-13 06:39:10
昆明医科大学学报(2021年1期)2021-02-07 01:06:50
生物化工(2021年2期)2021-01-19 21:28:13
生物化工(2020年1期)2020-02-17 17:17:58
读与写(2019年35期)2019-11-05 09:40:46
现代职业教育·高职高专(2018年7期)2018-05-14 16:20:40
兽医导刊(2016年6期)2016-05-17 03:50:50
医学研究杂志(2015年2期)2015-06-10 06:45:00
当代畜禽养殖业(2014年5期)2014-08-31 02:50:50
