
巨噬细胞铁超载在动脉粥样硬化中的研究进展*
2023-12-04 02:10:16巴静静于杨李元民
中国病理生理杂志 2023年11期
巴静静, 于杨, 李元民△
[1山东第一医科大学(山东省医学科学院),山东 济南 250117;2山东第一医科大学(山东省医学科学院)实验动物学院(省实验动物中心),山东 济南 250117;3山东第一医科大学第二附属医院心内科,山东 泰安 271000]
动脉粥样硬化性心血管疾病(atherosclerotic cardiovascular disease, ASCVD)是目前全球主要死因之一,据世界卫生组织报告估计,每年有1 790万人死于心血管疾病,占所有死亡人数的32%[1-2]。动脉粥样硬化(atherosclerosis, AS)是ASCVD最主要的病理学改变。它的发生和发展涉及脂质堆积、氧化应激、炎症以及血管和血细胞功能障碍,最终可能导致其临床并发症-诸如心肌梗死和中风等[3-5]。
巨噬细胞是AS病变中的主要免疫细胞,其增殖、聚集、衰老和死亡在AS的发生和发展中起关键作用[6]。在斑块内,巨噬细胞通过吞噬修饰的低密度脂蛋白(low density lipoprotein, LDL)形成泡沫细胞参与AS病变的进展[7]。铁是生理条件下参与巨噬细胞不同功能的必需矿物质。铁死亡是一种不同于细胞坏死和自噬的非凋亡性细胞死亡形式,已被证明通过多种信号通路参与AS的形成和发展[8]。铁死亡的主要特征是高水平的Fe2+通过Fenton反应产生活性氧(reactive oxygen species, ROS),造成脂质过氧化物积聚,从而引起氧化应激,继而导致DNA、蛋白质和脂质损伤[9]。巨噬细胞铁死亡在AS斑块的不稳定性中起着关键作用。巨噬细胞铁超载是铁死亡的直接原因,因此,调节巨噬细胞铁代谢可能是稳定斑块和抑制AS进展的重要途径[10]。本文从铁代谢的角度介绍了巨噬细胞铁超载和铁死亡对AS的影响。
1 巨噬细胞中的铁调节
铁是许多细胞和生物过程(包括氧运输和能量代谢)的重要调节剂。循环中铁主要以与转铁蛋白(transferrin, Tf)结合、血红蛋白和铁蛋白的形式存在,保护细胞免受铁诱导的氧化损伤。90%的铁通过网状内皮系统从衰老的红细胞中回收,用于骨髓造血,或者储存在肝脏中。
巨噬细胞摄取铁的主要途径是来自老化的红细胞或凋亡细胞以及转铁蛋白介导的摄取[11]。红细胞在ROS和水解酶作用下消化成血红素,血红素氧合酶1(heme oxygenase-1, HO-1)将血红素裂解成Fe2+、一氧化碳和胆绿素[12]。三价铁(Fe3+)与转铁蛋白结合形成Tf-Fe2,通过转铁蛋白受体1(transferrin receptor 1,TFR1)进入巨噬细胞,在内体被铁还原酶还原为Fe2+后,通过二价金属转运蛋白1(divalent metal transporter1, DMT1)储存在铁蛋白和的不稳定铁池(labile iron pool, LIP)中。大部分Fe2+被运输到线粒体,以协助电子传递链产生能量[13],过量的Fe2+通过铁转运蛋白(ferroportin, FPN)释放到细胞外[14]。Fe2+在胞外释放之前需要被具有铁氧合酶活性的CP蛋白氧化成Fe3+的形式[15](图1)。
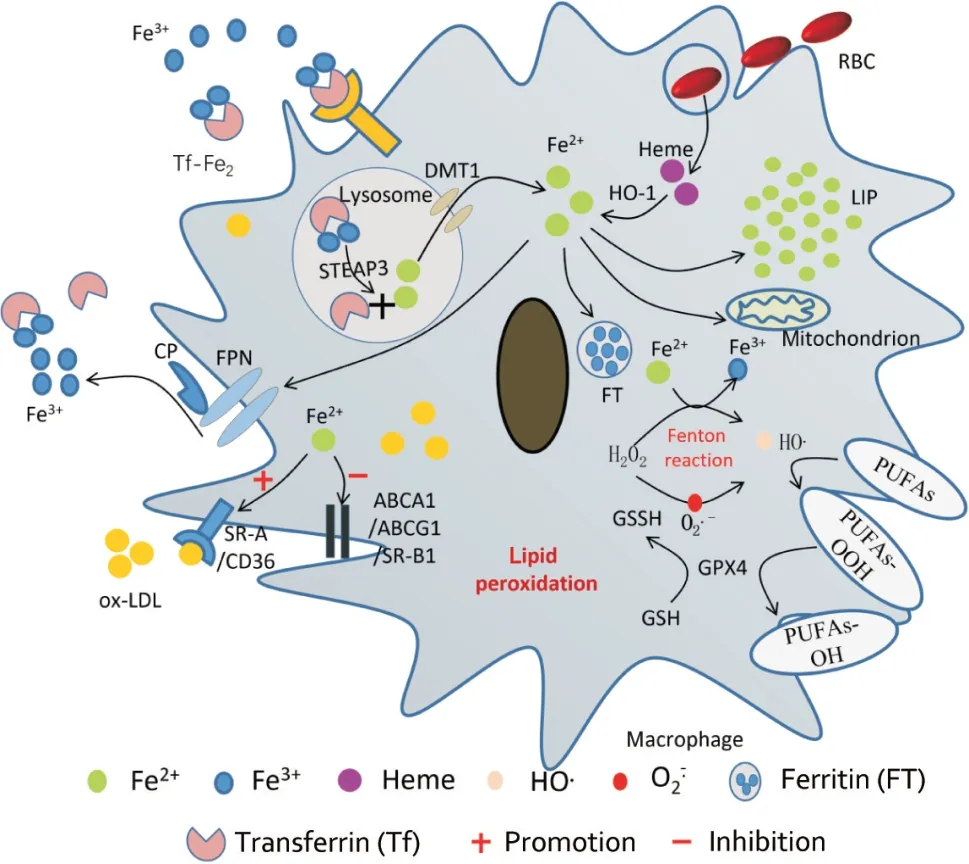
Figure 1. Iron metabolism of macrophages. The binding of Fe3+to transferrin (Tf) leads to the formation of Tf-Fe2,which subsequently enters macrophages through transferrin receptor 1 (TFR1). Iron reductase facilitates the reduction of Fe3+ to Fe2+ within the endosome, enabling its subsequent transport into the cytoplasm of macrophages via DMT1. Following phagocytosis by macrophages, the red blood cells are digested into Hb, driven by the concerted actions of reactive oxygen species (ROS) and hydrolase. The resultant breakdown products of Hb are then released into phagocytic vacuoles. Subsequently, Fe2+ is generated from heme under the action of heme oxygenase-1(HO-1). Fe2+ has four main pathways. It may be stored within the labile iron pool (LIP), transported to mitochondria for energy generation, stored as Fe3+ in ferritin (FT), or released to the extracellular environment through ferroportin (FPN). Prior to extracellular release, Fe2+ must be oxidized to Fe3+ by ceruloplasmin (CP), a protein with ferric oxidase activity. Iron overload provokes the upregulation of scavenger receptors, namely, SR-A1 and CD36, while simultaneously exerting a downregulating effect on cholesterol export proteins, namely, ABCA1 and ABCG1, resulting in lipid accumulation and consequent foam cell formation. Glutathione peroxidase 4(GPX4) requires glutathione (GSH) as a cofactor to catalyze the reduction of potentially toxic polyunsaturated fatty acid hydroperoxides (PUFAs-OOH) in the cell membrane to non-toxic polyunsaturated fatty acid hydroxides (PUFAs-OH).图1 巨噬细胞的铁代谢
全身铁稳态由各种铁加工组织和细胞(包括巨噬细胞、红细胞、肝细胞和十二指肠上皮细胞)的相互作用精密控制,并由铁调素-铁转运蛋白轴调节。FPN是唯一已知的铁输出蛋白[16]。细胞内和循环铁水平由铁调素维持,铁调素是由肝细胞产生,通过调节肠细胞、肝细胞和巨噬细胞表面的FPN量,从而调控循环中铁的浓度[17]。Ox-LDL和铁的沉积触发铁调素的自分泌,并加重AS。研究表明,铁可通过细胞因子IL-6和IL-1激活巨噬细胞,并上调铁调素的表达[18]。铁调素通过降解FPN增加巨噬细胞内的铁聚集,Fe2+通过参与Fenton反应提供电子促进脂质过氧化,产生ROS从而促进AS斑块进展[14]。此外,核受体共激活因子4(nuclear receptor coactivator 4,NCOA4)介导的铁自噬对维持细胞和全身铁稳态至关重要[19]。铁蛋白是细胞内的铁储存蛋白,NCOA4将铁蛋白转运到溶酶体或蛋白酶体进行降解,释放Fe2+。过度激活铁自噬将导致巨噬细胞的铁超载,从而减少谷胱甘肽过氧化物酶4(glutathione peroxidase 4, Gpx4)和谷胱甘肽(glutathione, GSH),最终引起巨噬细胞铁死亡[20]。
2 巨噬细胞铁超载可导致AS
铁是动脉粥样硬化的驱动因子,是一种能够引起脂质氧化和组织损伤的促氧化剂。研究表明,与正常动脉壁相比,AS病变中铁的含量增加,促进脂质过氧化,而铁螯合剂去铁胺可抑制这种作用[21]。在AS斑块中,铁、巨噬细胞、凋亡细胞共定位,表明巨噬细胞铁超载与细胞凋亡和AS有关[22]。
大量流行病学研究报告表明,血清铁浓度升高与动脉粥样硬化严重程度增加有关[23-24]。同时,实验证明通过静脉切开术、献血或应用铁螯合剂去铁胺等策略可降低动脉粥样硬化的风险[25-27]。已有研究表明,铁超载可通过氧自由基和脂质过氧化损伤血管内皮细胞,而铁死亡诱导剂Erastin引起的铁死亡相关蛋白Gpx4的下调和ACSL4的上调通过引起内皮细胞的铁死亡而加剧AS的发展[28]。此外,铁超载可通过促进AS中泡沫细胞的形成和炎症反应以及损伤平滑肌细胞而加剧AS斑块的形成和降低斑块的稳定性[29]。Luo等[30]研究显示,在铁含量为200 mg/kg的高脂饮食组动脉粥样硬化斑块中有大量的铁沉积,<20 mg/kg的高脂饮食组斑块中没有观察到铁沉积。研究表明,斑块中铁滞留通过诱导巨噬细胞泡沫化促进AS[18],而低铁饮食通过减少巨噬细胞和紊乱平滑肌细胞的聚集来改善AS。此外,铁络合剂去铁蛋白原抑制了AS饮食中ApoE-/-小鼠的斑块脂质氧化和斑块形成[31]。以上实验研究都说明了铁超载在促进AS过程中的重要作用。
3 巨噬细胞铁超载导致AS进展的机制
3.1 巨噬细胞铁超载导致铁死亡 铁死亡是依赖于细胞内铁浓度引起毒性脂质过氧化物蓄积的调节性细胞死亡形式[32]。铁死亡可能是GSH耗竭、铁超载和ROS过度生成共同作用的后果[14]。斑块内巨噬细胞清除红细胞后Fe2+含量增加,过量的Fe2+通过Fenton反应与H2O2相互作用,生成羟基自由基(andhydroxyl radicals, HO.),导致细胞内ROS累积以及细胞死亡。ROS包括超氧化物、H2O2和HO.[33]。细胞HO.将多不饱和脂肪酸(polyunsaturated fatty acid,PUFAs)氧化成PUFAs-OOH,脂质过氧化物的过量产生使细胞膜的流动性和通透性发生改变,导致巨噬细胞损伤以及铁死亡[34](图1)。铁沉积和脂质过氧化是人类AS斑块晚期的常见病理特征[35]。
巨噬细胞铁超载及其产生的ROS通过催化细胞膜中磷脂的氧化直接诱导细胞的铁死亡[8,36]。铁超载的巨噬细胞Fe2+通过FPN以Fe3+的形式释放到细胞外。此外,巨噬细胞铁死亡后细胞膜破裂,细胞内铁蛋白、Fe2+、ox-LDL以及促炎细胞因子释放到斑块中,诱发促炎反应[37],放大斑块中氧化应激环境,从而促进斑块的不稳定性[10,38]。斑块中的ROS可以从铁蛋白中释放铁[39],将Fe3+还原成Fe2+。Fe2+通过Fenton反应产生ROS,增加斑块内氧化应激,从而导致组织损伤(图2)。

Figure 2.Iron overload and ferroptosis of macrophages promote the progression of AS. The presence of Fe2+ within macrophages stimulates the production of reactive oxygen species(ROS) via Fenton reaction, thereby triggering lipid peroxidation and potential ferroptosis.Fe2+-induced oxidation of low-density lipoproteins (ox-LDL) promotes endothelial dysfunction, and stimulates an inflammatory response to recruit macrophages to the affected area. Following phagocytosis of ox-LDL by macrophages, Lipid deposition formed foam cells. Iron-overloaded macrophages exert a destabilizing influence on the plaques through up-regulating proteolytic enzymes. Within the atherosclerotic plaque, Fe2+ generates harmful ROS via Fenton reaction, which contributes to a pro-oxidative microenvironment within the plaque, and can stimulate smooth muscle cell (SMC) proliferation, migration as well asendothelial dysfunction-collectively promoting the progression of AS.图2 巨噬细胞铁超载与铁死亡促进AS进展
大量的ROS可导致巨噬细胞内氧化还原稳态失衡。GSH是体内主要的抗氧化剂[40],而Gpx4是唯一能够直接将细胞膜中有毒的PUFAs-OOH还原成无毒的PUFAs-OH,去除多不饱和脂肪酸的过氧化,同时将GSH转化为氧化型谷胱甘肽[41]。GSH的耗竭会导致Gpx4失活和脂质过氧化物的增加,最终导致铁死亡。因此,清除多余的铁和减少ROS的产生可能是未来治疗和预防AS的新策略。
3.2 巨噬细胞铁超载促进脂质沉积 在生理状态下,巨噬细胞中的游离胆固醇与胆固醇酯处于动态平衡。天然的LDL与LDL受体结合,内吞入巨噬细胞,胆固醇酯被溶酶体酶水解成游离胆固醇和游离脂肪酸,游离胆固醇通过转运蛋白运输到胞外。由于LDL受体随着细胞内胆固醇水平增高而减少,巨噬细胞摄取LDL不会引起胆固醇酯在细胞内堆积[42]。
Fe2+通过Fenton反应产生的ROS将LDL氧化为ox-LDL,导致内皮细胞功能障碍,同时诱导炎症反应募集巨噬细胞[43]。Ox-LDL主要通过清道夫受体依赖的吞噬和胞饮作用进入巨噬细胞内,清道夫受体包括SR-A1、CD36和LOX-1[44],其表达不受细胞内胆固醇水平的负反馈调节。
巨噬细胞不受限制的吞噬ox-LDL,导致胆固醇的摄取和外排的动态平衡失衡,后脂质沉积于细胞内形成泡沫细胞[45]。Ox-LDL在巨噬细胞溶酶体中被水解成游离胆固醇和游离脂肪酸,过多的游离胆固醇被乙酰辅酶A乙酰转移酶1(acetyl coenzyme a acetyltransferase1, ACAT1)转化为胆固醇酯,胆固醇酯以脂滴的形式储存在细胞中。中性胆固醇酯水解酶(nCEH和NCEH1)能将胆固醇酯水解成游离胆固醇,然后通过胆固醇转运蛋白ATP结合盒(ABC)A1介导胆固醇流出至载脂蛋白A1(ApoA1)[46],通过ABCG1和SR-BI介导胆固醇流出至HDL[47]。
铁超载的巨噬细胞清道夫受体SR-A1和CD36表达增加,促进其吞噬ox-LDL[48],此外,胆固醇输出者(ABCA1和ABCG1)在铁超载的巨噬细胞中受到抑制,导致脂质堆积形成泡沫细胞,泡沫细胞的聚集成为AS斑块的脂质核心[49](图1)。铁超载的巨噬细胞通过上调蛋白水解酶,释放MMP降解细胞外基质,导致斑块稳定性下降[37]。此外,炎症刺激可上调清道夫受体与ACAT1的表达,抑制胆固醇转运蛋白ABCA1和ABCG1以及NCEH的表达,增加脂质摄取,而胆固醇逆转运途径受限,导致游离和酯化胆固醇在巨噬细胞中沉积并产生泡沫细胞[50-51]。
因此,铁超载导致的巨噬细胞中胆固醇的积累是促进AS斑块生长的关键过程,也是决定斑块稳定性的主要因素[52]。
3.3 巨噬细胞铁超载促进其极化 巨噬细胞可以分化为M1或M2表型,M1在斑块进展中占主导地位,M2在斑块消退中发挥关键作用[53]。M1是脂核中的主要炎性巨噬细胞群[54]。在AS病变中,巨噬细胞在胆固醇晶体、促炎细胞因子和ox-LDL等作用下极化成M1型巨噬细胞[55],通过分泌促炎细胞因子如TNF-α、IL-1和IL-6等,加速AS的形成和发展[56-57]。此外,M1巨噬细胞可通过释放MMP降解纤维帽内的细胞外基质,降低斑块的稳定性。
M2巨噬细胞在细胞因子IL-4和IL-13的作用下被极化,并分泌抗炎细胞因子,如IL-10、转化生长因子β和胶原[58-59]。与M1型相比,M2型巨噬细胞表达更多数量的清道夫受体(如CD36)[60],通过吞噬作用清除凋亡或死亡的细胞,并产生胶原蛋白以增厚纤维帽,从而稳定斑块[61]。除M1和M2表型外,斑块中还观察到其他极化的细胞,如M(Hb)、MHEM、Mox和M4[7]。M(Hb)和MHEM型与M2型巨噬细胞一样具有抗炎作用,可产生IL-10等抗炎细胞因子,从而阻止斑块进展。Mox和M4型产生IL-6和TNF-α等促炎细胞因子,促进AS的进展[62-63]。巨噬细胞的不同表型对AS的形成有着不同的作用,将巨噬细胞由促炎转化为抗炎表型有助于抗AS。
铁超载不仅可以通过诱导巨噬细胞积聚和炎症反应促进动脉粥样硬化,还可以通过增强糖酵解诱导巨噬细胞向M1型极化[64-66]。细胞内铁超载能够增加M1巨噬细胞的数量并诱导M2型巨噬细胞向 M1表型转换[67]。M1巨噬细胞表达高水平的铁蛋白和低水平的FPN,铁输出减少,导致巨噬细胞内的铁含量增加。细胞内铁蓄积可通过上调CD36等清道夫受体增加ox-LDL的摄入,促进泡沫细胞形成,并通过TLR-4途径激活促炎的M1巨噬细胞[68]。M1巨噬细胞具有强大的吞噬活性并分泌促炎细胞因子,如TNF-α[69]。长期被炎症因子持续渗透的血管容易发生LDL聚集和氧化,以及AS斑块的铁沉积,促进AS斑块中巨噬细胞的铁死亡[5]。总之,铁超载可通过促进ROS的产生、炎症反应、泡沫细胞的形成和巨噬细胞极化改变巨噬细胞功能。
3.4 巨噬细胞铁超载参与内皮细胞和血管平滑肌细胞激活促进AS 血管内皮细胞是铁超载损伤的主要靶细胞之一,铁超载通过其促炎和促氧化作用导致内皮细胞功能障碍,直接导致AS进展[70]。Fe2+诱导产生的ox-LDL通过激活炎性转录因子NF-κB促进内皮功能障碍[43]。受NF-κB调控的靶基因包括血管黏附分子-1、细胞间黏附分子-1及单核细胞趋化蛋白-1等,这些细胞因子均参加单核细胞的浸润和迁移[29]。此外,铁超载通过上调促炎细胞因子的表达降低内皮型一氧化氮合酶的活性,从而降低血管内皮的舒张功能[71]。总之,铁超载能通过增加氧化应激、降低NO生物利用度、促进炎症反应等导致内皮细胞受损而推动AS进展[72]。
巨噬细胞铁超载引起斑块中铁含量增加,Fe2+通过Fenton反应产生ROS,增加斑块促氧化微环境,可引起SMCs增殖、凋亡、表型转换和钙化等功能障碍[73]。铁超载促进巨噬细胞向M1型极化,M1型巨噬细胞通过产生转化生长因子-β或血小板源生长因子促进SMCs增殖以及向血管内膜迁移。SMCs可分化为巨噬细胞样SMCs吞噬脂质后形成泡沫细胞,从而降低斑块稳定性并推动AS的发展[74]。此外,铁刺激还可以通过激活BMP2和IL-24途径诱导SMCs 钙化[72]。铁超载以及各种因素的刺激会使SMCs异常增殖、钙化,最终导致血管管腔狭窄。
4 总结与展望
铁死亡是一种由铁依赖性氧化还原失衡引起的调节性细胞死亡方式。许多研究证明铁超载通过促进炎症反应、ROS产生、巨噬细胞极化和泡沫细胞形成来改变巨噬细胞的功能,从而促进AS斑块的形成。此外,铁超载还可导致内皮细胞功能障碍,破坏SMCs,刺激免疫细胞聚集,加速AS的进展。因此,在AS的诊断和治疗中需要考虑铁对AS的影响,这表明铁死亡可以成为治疗心血管疾病的潜在靶点。鉴于大多数心血管疾病是慢性的,心血管疾病的药物调节应长期使用,需要关注用药安全性;据报道,一些铁死亡抑制剂如去铁胺具有耳毒性和神经毒性[75]。此外,铁死亡诱导剂能有效地抑制癌症[76-77],使用铁死亡抑制剂治疗心血管疾病时,可能会增加癌症发生的风险。因此,需要深度探讨巨噬细胞铁死亡在心脑血管疾病中的作用及机制,以期为ASCVD患者提供治疗新靶点。
猜你喜欢
科学(2022年4期)2022-10-25 02:43:32
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
中成药(2018年9期)2018-10-09 07:18:36
中成药(2018年1期)2018-02-02 07:19:53
中成药(2017年4期)2017-05-17 06:09:26
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
天津科技大学学报(2015年3期)2015-04-16 04:54:59
云南中医学院学报(2014年5期)2014-07-31 18:00:10
食品工业科技(2014年15期)2014-03-11 18:17:43
