
α-Fe中氢与碳化物相互作用的第一性原理研究
2023-11-28 09:56黄剑新
上海金属 2023年6期
黄剑新 王 昊
(上海大学 材料科学与工程学院,上海 200444)
钢在工业应用中的扩张在提高强度要求和微观结构复杂性以及钢对氢脆(hydrogen embrittlement,HE)的敏感性之间遇到了两难境地[1-2]。到目前为止,许多研究试图探索HE 机制,并为提高抗氢脆性能提供了方向[3-4]。研究得出[5-6],基体中的H 原子会扩散并累积到应变区域和晶体缺陷处,这会增加位错的迁移率或削弱晶界的结合强度,最终导致材料断裂。先进高强度钢在生产和使用中不可避免地接触到H,即使H浓度只有几μg/g 也会引起材料性能恶化,因此提高抗氢脆性能的一种方法是引入足够量的有效H陷阱。
研究发现,虽然钢基体中的空位和位错可以捕获H 原子,但与H 原子的结合较弱,导致H 容易脱离,称为可逆陷阱[7-8]。众多研究表明,钢中均匀分布的碳化物沉淀如TiC、VC 和NbC 不仅可以诱导沉淀强化[9-11],而且可以提高钢的抗氢脆性能[12-15]。此外,观察发现,大多数H 原子位于片状沉淀物的扩展基底界面上[16-17]。因此推测,抗氢脆性能的提高归因于析出物内部或析出物与钢基体之间界面的捕获位点。然而,对于H 原子的特定捕获位点仍有待更深入的研究。
Takahashi等通过原子探针层析(atom probe tomography,APT)发现,VC 沉淀的失配位错核心[17]和VC 沉淀界面上的C 空位[18]是主要的H陷阱位点;其中,通过热解吸光谱(thermal desorption spectroscopy,TDS)测得C 空位的捕获能为60 kJ/mol。Turk等[19]的研究表明,VC 捕获H 的能力取决于其有效表面积。而Wei 和Tsuzaki[14]将捕获的H 原子量与沉淀物的体积相关联,发现与它们的有效表面积无关。Shi 等[20]通过TDS测得了NbC 对H 的捕获能为81.8 kJ/mol,而Wallaert等[15]报道的捕获能为39 ~68 kJ/mol。Wei等[21]通过TDS研究发现,3 种具有半共格界面的碳化物捕获H 原子的能力大小顺序为NbC >TiC >VC。采用APT虽然可以观察碳化物捕获H的行为,但不能提供具体捕获位点的直接证据。此外,通过TDS对H捕获能的研究只能提供间接能量信息[22-23]。
为解决上述问题,采用原子模拟方法可以深入了解H 扩散和捕获的微观机制。基于密度泛函理论(density functional theory,DFT)的第一性原理计算是一种非常可靠的方法,可应用于不同的材料。Ma等[24]使用第一性原理计算了H在有或没有C 空位的各种碳化物和氮化物中的溶解能。Shi等[20]模拟了NbC/Fe 界面的Kurdjumov-Sachs 取向并计算了其H 捕获能。Di Stefano等[25]模拟了不同的TiC/Fe界面(共格、半共格和非共格)并计算了可能捕获H 原子的多个位置,并推测由于岩盐结构相同,TiC、VC 和NbC 应该具有相似的结论。然而目前对这3 种碳化物的捕获位置和性质的定量研究还比较滞后,相关数据还比较缺乏。
本文对α-Fe 基体(bcc)与上述3 种碳化物(沉淀)的界面进行建模,并通过第一性原理研究H陷阱特性,根据计算结果获得了H 在相应捕获位点的偏析能。
1 计算方法
1.1 理论方法
通过大量试验可知,具有岩盐状面心立方结构的碳化物MC(M =Ti,V,Nb)与bcc-Fe 基体存在3 种不同的界面:共格、半共格和非共格。随着MC尺寸的增大,其与基体之间的共格性也逐渐消失,界面由共格转变为非共格,失配位错出现在(001)Fe/(001)TiC平面[26-27]。其中,共格和半共格界面具有(001)Fe/(001)TiC和[001]Fe/[001]TiC的Baker-Nutting(B-N)取向关系[14]。然而,对于非共格界面,目前还没有相关试验得出明确的取向关系,因此本文不考虑非共格界面。
共格界面的特点是界面上原子平面的完美重合,因此可以用小型超胞模型表示该系统。而在图1 所示的半共格界面情况下,累积的弹性应力通过形成失配位错来释放,失配位错之间的间距通常为几纳米,导致模型过大。但是半共格界面可近似地分成宽的共格区域,这些区域被包含失配位错核心的相对狭窄区域周期性地打断[25],其中的长程应变场(实线和虚线之间的区域)在该近似中可被忽略。

图1 半共格界面示意图(实线是位错核心,实线和虚线之间的区域表示失配程度逐渐增加;A区域是完美共格界面,B区域是失配位错的核心,C区域是两个垂直位错的交结处)Fig.1 Schematic diagram of the semi-coherent interface(the solid line is the dislocation core,and the area between the solid line and the dashed line indicates that the degree of mismatch increases gradually;region A is the perfect coherent interface,region B is the core of the misfit dislocation,and region C is the junction of two vertical dislocations)
1.2 计算细节
本文所有DFT 计算都在从头计算软件包VASP(Vienna Ab-initio Simulation Package)[28-29]中完成。投影增强波(projector-augmented wave,PAW)方法用于描述离子和电子之间的相互作用[30],交换关联泛函由广义梯度近似(generalized gradient approximation,GGA)[31]表示。考虑到系统的磁性,所有计算均使用自旋极化进行。电子自洽能量和原子力收敛标准分别设置为10-5eV/atom和10-2eV/Å。对不同构型进行布里渊区k点和平面波截断能的收敛测试。
块体Fe或MC晶体内的孤立间隙H 原子的溶解能被定义为:
2 结果与讨论
计算的晶格常数、H 在各块体相中的溶解能和迁移能垒如表1所示,与其他研究结果非常一致。bcc-Fe基体中氢最稳定的间隙位点是四面体位点[29,32],2 个最近的四面体位点构成H 原子的扩散路径。在完美化学计量的TiC 中,H 原子最稳定的位置是“三角”位点,其中H 原子被{111}平面的3 个Ti 原子包围,而不是位于由4个Ti和4 个C交替占据的立方体中心[25]。
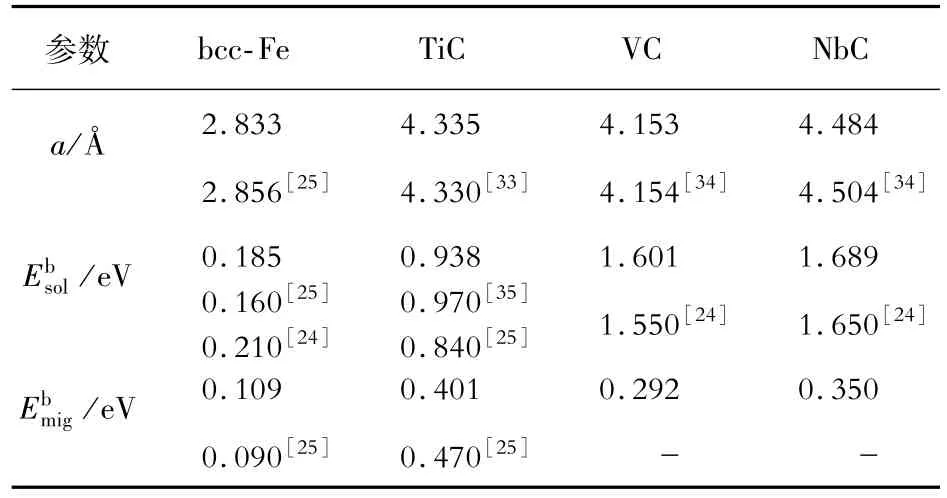
表1 各块体相中H原子的晶格常数、溶解能和迁移能垒汇总Table 1 Summary of the lattice constants,solution energy and migration energy of H atom in each bulk phase
沿B-N 方向,bcc-Fe 与TiC(8%,晶格失配度,下同)、VC(4%)和NbV(11%)之间存在较小程度的失配,这主要是通过将bcc-Fe 的初始横向晶格常数设置为MC 的横向晶格常数来调节的,因为MC是较硬的相。MC/Fe界面的共格部分对应于Fe-on-C 构型,是3 种构型中最稳定的界面[25]。此外,额外的几何自由度为两个晶体平行和垂直于界面平面的相对平移。Fe 在半共格界面处的错配位错核心中心沿< 110 > Fe 或<10 >Fe方向相对于MC偏移半个晶面间距,产生Bridge构型,因此类似于失配位错核[14]。此外,当沿〈110〉Fe 和<10 >Fe 的两个垂直失配位错(如图1 所示)相交时,在位错核的交叉处形成Fe-on-Ti构型。图2 列出了3 种不同类型的界面构型,分别表示为“Fe-on-C”“Bridge”和“Feon-M”。横向使用了2 ×2 超胞,而图2(b)中界面垂直方向的Fe 和MC 中的5 个原子层已经过测试,足以保证计算的准确性。
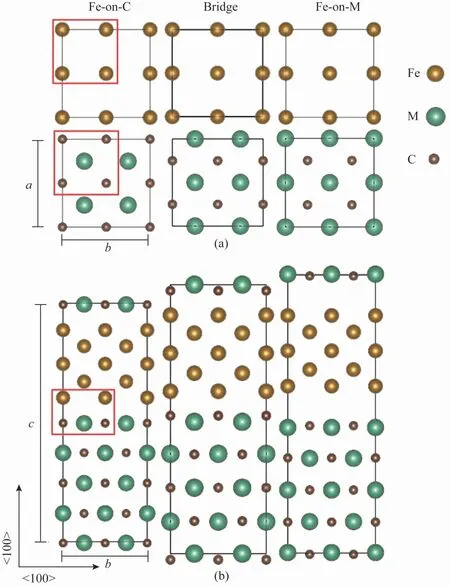
图2 3 种不同类型的界面构型(顶部和底部面板分别表示(001)Fe和(001)TiC界面平面(a)、(001)Fe/(001)TiC界面3 种构型的原子结构(b))Fig.2 Three different types of interface configurations(top and bottom panels represent the(001)Fe and(001)TiC interface planes(a)and atomic structures of the three investigated configurations for the(001)Fe/(001)TiC interface(b),respectivly)
构建的超胞完全弛豫,优化后的参数和界面能如表2 所示。可见模型的横向晶格常数与块体MC的横向晶格常数接近,进而证实了主要通过拉伸Fe相来解决晶格失配。界面能的计算结果与其他研究非常一致。Fe-on-C 对应于图1 中的共格界面区域A;Bridge 和Fe-on-M 对应于图1中的区域B和C,即失配位错的核心和交结。文献[36]中的3 种不同构型使用了相同的晶格常数(普通晶格)。

表2 MC/Fe界面超胞尺寸(a、b、c)、Fe和Ti在<001 >方向上的界面距离d,以及本文计算的界面能γ和其他工作中γrefTable 2 Supercell dimensions(a,b,c)of the MC/Fe interfaces,the interface distance d corresponding to the distance between Fe and Ti in the <001 >direction,and the interface energies obtained in this work(γ)and other works(γref)
基于上述3 种不同的界面构型,H 原子最初被放置在界面中不同的高对称位置并进行了优化。图3 为界面处可能的H 原子陷阱位点,计算得到的H 原子偏析能为负值。由于模型的对称性,只需取界面处横向宽度的1/4(即图2 中红框所示区域)用以表征H捕获位点。结果发现大部分位点并不在某个多面体的中心,而是在靠近界面的Fe原子平面上,且保持初始的对称性。

图3 3 种不同构型中稳定位置(红色多边形)的示意图(仅显示了图2 中红色框所示区域,黑色、灰色和白色圆圈分别代表Fe、C和Ti原子)Fig.3 Schematic representations of the stable positions(red polygons)in three different configurations(the area indicated by the red box in Fig.2 is shown only;black,grey and white circles represent Fe,C and Ti atoms,respectively)
计算的H 原子在不同捕获位点的偏析能如表3 所示。可以发现:在Fe-on-C 构型中,H 在C1 和C3 位点更有利,TiC/Fe 和NbC/Fe 比VC/Fe更利于捕获H;在Bridge 构型中,对于所有MC/Fe,H最稳定的位置是B2 位点。此外,NbC/Fe和TiC/Fe 比VC/Fe 更利于捕获H;对于Feon-M构型,H在大多数位点的偏析能较小,只在少数情况下偏析能较大,为0.014 eV。除了VC/Fe中的Bridge 和NbC/Fe 中的Fe-on-C 构型外(其有利位点在Fe 相内),各构型中最稳定的H陷阱位点都在Fe 平面上。失配位错核心及其交结的捕获能力强于共格界面,这与大多数研究结论一致。此外,MC/Fe 半共格界面处碳化物捕获H能力的大小顺序为NbC >TiC >VC,这也与Wei等[21]的结论一致。

表3 计算的纯(001)Fe/(001)TiC界面处不同稳定位点处H原子的偏析能(位点标签为图3 中所示位置)Table 3 Calculated segregation energy of H atom at different stable sites in the pure(001)Fe/(001)TiC interface(the labels for the sites refer to the location shown in Fig.3)
3 结论
本文详细研究了H 原子与bcc-Fe 中不同碳化物之间的相互作用。MC/Fe 的共格、半共格(失配位错核和位错核的交结)界面构型有“Feon-C”“Bridge”和“Fe-on-M”3 种。H原子的捕获位置多在靠近界面的Fe 原子层或Fe 基体内部;MC/Fe半共格界面碳化物捕获H 能力的大小顺序为NbC >TiC >VC。
猜你喜欢
上海金属(2022年6期)2022-11-25
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
数学物理学报(2019年5期)2019-11-29
模具制造(2019年3期)2019-06-06
当代陕西(2019年6期)2019-04-17
数学物理学报(2017年5期)2017-11-23
上海金属(2016年4期)2016-11-23
潍坊学院学报(2016年6期)2016-04-18
长江大学学报(自科版)(2014年1期)2014-03-20
无机化学学报(2014年4期)2014-02-28
