
宁夏荒漠草原土壤细菌与真菌群落对降水变化的响应
2023-11-22 09:38米扬郭蓉王媛王占军蒋齐俞鸿千马琨
草业学报 2023年11期
米扬,郭蓉,王媛,王占军,蒋齐,俞鸿千,马琨*
(1. 宁夏大学西北土地退化与生态恢复国家重点实验室培育基地, 宁夏 银川 750021;2. 西北退化生态系统恢复与重建教育部重点实验室, 宁夏 银川 750021;3. 宁夏农林科学院林业与草地生态研究所,宁夏 银川 750001)
联合国政府间气候变化专门委员会(Intergovernmental Panel on Climate Change, IPCC)报告指出,全球气候变化背景下会有更多地区遭受频发干旱所带来的一系列问题[1]。与其他类型草原相比,降水变化对荒漠草原生态系统的影响更为显著[2],这是因为该类型的生态系统很容易受到气候及年降水量变化的影响[3]。土壤微生物作为生态系统的重要组分,具有重要的生态功能[4]。细菌和真菌作为主要土壤微生物类群,广泛参与气候、生物及土壤特性变化及其相互作用的过程[5],能够敏锐地感知土壤生态系统的各种细微变化[6],对土壤养分获取(如氮、磷)和碳循环等生态系统过程影响重大[7-10]。
降水会通过对土壤生物和非生物因素的影响改变微生物群落组成[11],通过改变土壤水分有效性、调节半干旱草原的植物生产、碳分配,影响微生物生长[12]。有研究证实,降水可通过影响植被生长和养分吸收改变植被群落组成[9],但不同植被对于微生物群落组成和多样性的影响结果却不尽相同[13]。降水通过改变土壤水分有效性影响微生物的生理策略,使其能够耐受动态水势环境变化[14]。为了应对干旱胁迫,微生物进化出了渗透调节、休眠和生产胞外聚合物等多种生理策略[2]。微生物通过积累溶质(渗透物)保留细胞膨压,从而保证其能够在较低的水势环境下生存[15]。土壤微生物可能只是在缺水状态下休眠,当水分恢复时解除休眠。微生物的另一种生理策略是通过产生胞外聚合物从而在低基质电位下保持水分[2]。此外,土壤放线菌和酸杆菌等细菌类群有保持活性并在干旱条件下休眠的能力,因而可以在干旱的土壤环境中持续存在[2];而球囊菌等真菌类群可以通过菌丝连接应对土壤缺水状况,从而获取土壤中的营养物质,维系自身的存活[16]。
荒漠草地生态系统作为宁夏重要的天然屏障及重要生态资源,对西北地区的生态安全影响巨大[17]。宁夏荒漠草原区域是我国西北生态脆弱区[18],区域降水资源分配不均,在气候变化等各种因素的共同影响下,草地生态系统退化现象仍较严重。土壤微生物群落组成与单独的生物因素(如植物群落组成)及非生物因素(如土壤氮、磷、有机质)的相关关系已有较多研究[5,12,19-20];然而,有关降水变化作用下,土壤细菌和真菌群落与生物和非生物因子间关系的研究,还有待于进一步加强。因此,通过分析降水变化对宁夏荒漠草原土壤微生物群落及多样性的影响;利用结构方程模型,揭示不同土壤微生物类群对降水变化的响应机制,能为未来降水变化趋势预测,维护荒漠草原生态系统的稳定性提供理论依据。
1 材料与方法
1.1 研究区概况
研究区位于宁夏中部和北部的荒漠草原,属温带大陆性季风气候。荒漠草原植被群落组成以豆科、禾本科和菊科为主,主要包括甘草(Glycyrrhiza uralensis)、白草(Pennisetum centrasiaticum)、短花针茅(Stipa breviflora)、黑沙蒿(Artemisia ordosica)、猪毛蒿(Artemisia scoparia)和牛枝子(Lespedeza potaninii)等植物[21],土壤类型为灰钙土和风沙土。研究选取4 个具有不同降水特征的荒漠草原国家监测点作为观测样地,观测样地自2003 年开始围封管理。各荒漠草原监测区的年降水量主要分布在7-8 月,其降水量占年降水量的46.87%~64.40%。荒漠草原样地(T0)位于宁夏盐池县高沙窝镇(E 107.05°,N 38.08°),年均气温8.3 ℃,海拔1463 m,多年平均降水量231 mm,其优势种群为白草和甘草群落;样地(T1)位于宁夏红寺堡区太阳山(E 106.48°,N 37.44°),年均气温8.4 ℃,海拔1371 m,多年平均降水量154 mm,优势种群为短花针茅、牛枝子和猪毛蒿群落;样地(T2)位于宁夏灵武市白土岗新火村(E 106.62°,N 37.76°),年均气温8.8 ℃,海拔1340 m,多年平均降水量为137 mm,其优势种群为黑沙蒿群落;样地(T3)位于宁夏中卫市中宁县新堡创业村(E 105.73°,N 37.40°),海拔1377 m,多年平均降水量为114 mm,其优势植被种群为短花针茅群落。
1.2 试验设计
1.2.1 样地设置、植被调查与土壤采集 2018 年8 月在每个观测样地内(100 m×100 m)随机选取6 个样方(1m×1 m)进行植被调查,调查指标包括多度、盖度、高度、频度和生物量,植被群落多样性指数及丰富度指数的计算方法及主要结果见前期相关研究[21]。植被调查完成后,在每个样方内利用5 点采样法采集0~20 cm 土层的土壤样品,混合均匀,各土壤样本保持独立,共采集24 个土壤样本。通过低温保存箱带回实验室。土壤样本去除植物枯落物及植物根系后采用四分法混合均匀。随后,部分土壤样本过1 mm 筛后保存在-80 ℃进行高通量测序,剩余土壤样本待自然风干后进行土壤基础理化性质测定。土壤基础理化性状结果见前期相关研究[21]。
1.2.2 测定方法 土壤全氮(total nitrogen, TN)采用半微量凯氏法测定;有机质(organic matter, OM)采用重铬酸钾氧化外加热法测定; 全磷(total phosphorus, TP)采用HCIO4-H2SO4消煮、钼锑抗比色法测定;碱解氮(alkali-hydrolyzable nitrogen, AN)采用碱解扩散法测定;速效磷(available phosphorus, AP)采用NaHCO3浸提-钼锑抗比色法测定;速效钾(available potassium, AK)采用NH4OAC 浸提-火焰光度法测定;pH 采用酸度计(HI2221,欧洲)电位法测定(水土比为5∶1)[22]。
土壤细菌和真菌DNA 提取采用Fast DNA Spin kit(MP Biomedicals, Santa Ana, CA,美国)试剂盒,按照说明书步骤进行。细菌选择16Sr 基因的V3-V4 区段扩增,正向引物为338F(5′-ACTCCTACGGGACGGCA GCA-3′),反向引物为806R(5′-GGACTACGGGTATCTAAT-3′)。扩增参数:98 ℃预变性2 min;98 ℃变性15s,55 ℃退火30 s,72 ℃延伸30 s,最后72 ℃延伸5 min,30 个循环。扩增体系为25 μL,5×反应缓冲液5 μL,5×GC缓冲液5 μL,dNTP(2.5 mmol·L-1)2 μL,前引物1 μL(10 μmol·L-1),后引物1 μL(10 μmol·L-1),DNA 模板 40 ng,Q5 DNA 聚合酶(NEB 公司)0.25 μL,超纯水(ddH2O)补足至 25 μL。
采用特异引物(ITS1F/2043R)对土壤真菌基因的ITS1F-ITS2 区段进行扩增,正向引物为ITS1F(5′-CTTGGTCATTTAGAGGAAGTAA-3′),反向引物为2043R(5′-GCTGCGTTCTTCATCGATGC-3′)。PCR 反应体系为:5×Q5 缓冲液5.0 μL,5×Q5 高保真GC 增强剂5.0 μL,dNTPs 2.0 μL(2.5 mmol·L-1),DNA模板2.0 μL(0.1 ng·μL-1),正向引物和反向引物各1.0 μL(10 μmol·L-1),Q5 聚合酶0.25 μL(5 U·μL-1),灭菌超纯水8.75 μL,超纯水(ddH2O)补足至 25 μL。扩增条件:98 ℃预变性2 min,98 ℃变性15 s,55 ℃退火30 s,72 ℃延伸30 s,共30 个循环。
扩增产物使用浓度为2%的琼脂糖凝胶进行电泳检测。利用TruSeq® DNA PCR-Free Sample Preparation Kit 建库试剂盒构建文库,文库质检合格后使用IonS5TMXL 测序平台,利用单端测序(Single-End)的方法,构建小片段文库进行单端测序。根据Barcode 序列和PCR 扩增引物序列从下机数据中拆分出各样本数据,去除Barcode和引物序列后使用FLASH(V 1.2.7,http://ccb.jhu.edu/software/FLASH/)[23]对每个样本的reads 进行拼接,得到的拼接序列为原始Tags 数据(raw tags),通过严格的过滤处理[24]后得到高质量的Tags 数据(clean tags)。参照Qiime(V 1.9.1,http://qiime. org/scripts/split_libraries_fastq. html)[25]的Tags 质量控制流程,质控包括两部分:Tags 截取和Tags 长度过滤,Tags 截取是将raw tags 从连续低质量值(默认质量阈值为≤19)碱基数达到设定长度(默认长度值为3)的第一个低质量碱基位点截断;Tags 长度过滤是指将Tags 经过截取后得到的Tags 数据集,进一步过滤掉其中连续高质量碱基长度小于长度75%的Tags。经过质控处理后得到的Tags 再去除嵌合体序列,Tags 序列通过(https://github.com/torognes/vsearch/)[26]与物种注释数据库进行比对检测嵌合体序列,并最终去除其中的嵌合体序列[27],得到最终的有效数据。利用Uparse 软件(Uparse v 7.0.1001,http://www.drive5.com/uparse/)[28]对所有样本的全部 Effective Tags 进行聚类,以97%的一致性(identity)将序列聚类成为操作分类单元(operational taxonomic units,OTUs),同时会选取OTUs 的代表性序列,依据其算法原则,筛选在OTUs 中出现频数最高的序列作为OTUs 的代表序列。对OTUs 序列进行物种注释,用Mothur 方法与SILVA 132(http://www.arb-silva.de/)的SSUrRNA 数据库进行细菌物种注释分析(设定阈值为0.8~1.0),用Qiime 软件(Version 1.9.1)中的blast 方法(http://qiime. org/scripts/assign_taxonomy. html)[29]与Unit(v 7.2)数据库(https://unite. ut. ee/)[30]进行真菌物种注释分析。测序工作委托天津诺禾致源生物信息科技有限公司通过IonS5TMXL 测序平台进行单端测序。
1.3 数据分析
利用Excel 2010 进行数据整理,使用Qiime 软件(Version 1.9.1)计算微生物ACE、Chao1,Shannon 和Simpson 指数,而后使用R 软件进行α 多样性指数组间差异分析。通过非度量多维尺度分析(non-metric multidimensional scaling analysis, NMDS)和相似度分析检验(analysis of similarities, ANOSIM)土壤细菌和真菌群落间的β 多样性差异,基于 999 个排列的ANOSIM 计算与NMDS 相关的r值和P值。采用单因素方差分析(Duncan 多重比较法)检验不同处理间土壤细菌及真菌群落门水平间的显著性差异(P<0.05)。利用蒙特卡罗检验(Monte Carlo test)进行植被、土壤理化性状和微生物群落之间的冗余分析(redundancy analysis, RDA)。通过AMOS 软件构建结构方程模型,采用Origin 进行绘图。
2 结果与分析
2.1 土壤细菌和真菌可操作分类单元数及其分布特征
高通量测序结果表明(图1),T0、T1、T2和T3降水梯度下的土壤细菌总OTUs 分别为5130、6355、6149 和5494,特有OTUs 分别为235、564、496 和382。T0、T1、T2和T3降水梯度下的土壤真菌总OTUs 分别为2063、2492、2481 和2118,特有OTUs 分别为315、436、533 和286。土壤细菌群落总OTUs、真菌群落总OTUs 在各个降水梯度下具有相似的变化规律;均表现为随降水减少,呈先升高后降低趋势。每种降水梯度下土壤细菌总OTUs均为真菌总OTUs 的2.5~2.6 倍。
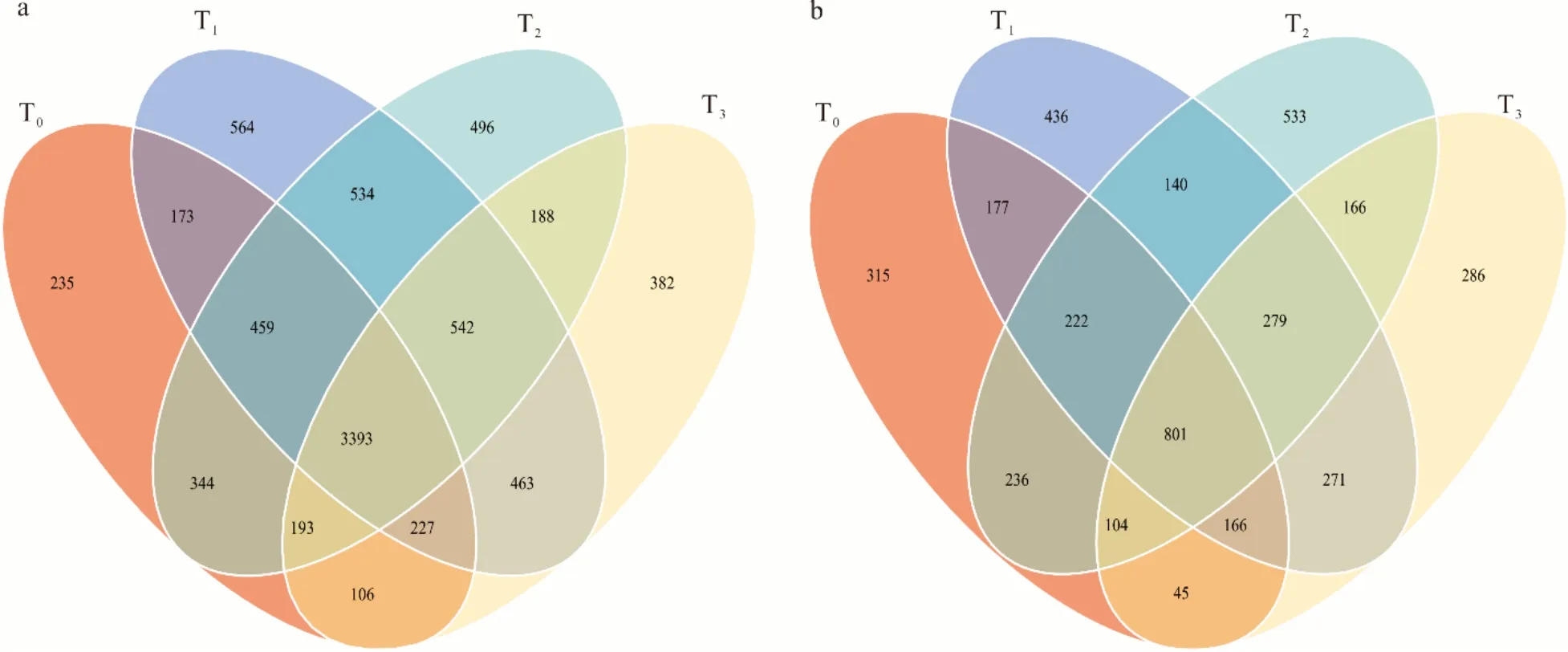
图1 土壤细菌(a)和真菌(b)独有和共有OTUs 数量分布的韦恩图Fig.1 Venn diagram of the number of shared and unique bacterial (a) and fungal (b) OTUs in the different treatments
2.2 土壤细菌和真菌群落相对丰度及优势种变化特征
不同降水量下荒漠草原观测样地基于门水平分类组成的土壤微生物物种差异分析结果表明(图2):无论降水如何变化,土壤细菌优势菌门始终为变形菌门(Proteobacteria)(13.55%~16.81%)、放线菌门(Actinobacteria)(14.57%~16.39%)和酸杆菌门(Acidobacteria)(8.63%~10.79%)(图2a)。土壤变形菌门相对丰度整体上表现出随降水减少呈降低的趋势;其中T0处理下变形菌门的相对丰度极显著高于其他处理。此外,土壤酸杆菌门和绿弯菌门(Chloroflexi)相对丰度在不同自然降水梯度下也产生显著差异(P=0.003),二者相对丰度随降水量减少呈显著升高趋势。

图2 土壤细菌(a)和真菌(b)群落在门水平上相对丰度差异Fig.2 Differences in the relative abundance of soil bacterial (a) and fungal (b) communities on the phylum level
真菌群落的优势类群为子囊菌门(Ascomycota)(15.89%~17.87%)和担子菌门(Basidiomycota)(7.55%~10.04%)(图2b)。子囊菌门相对丰度整体上随着降水量下降呈降低趋势,但在不同降水量变化下其相对丰度间无显著性差异;而担子菌门的变化趋势恰好与之相反,其相对丰度整体上随着降水量下降呈增加趋势。此外,非优势菌门的球囊菌门(Glomeromycota)相对丰度在不同降水观测样地间却存在极显著性差异。
2.3 不同降水梯度下荒漠草原土壤细菌及真菌群落α 多样性变化特征
由表1 可知,T0(231 mm)处理下细菌群落的Chao1 指数、ACE 指数(P=0.011)和香农-维纳指数显著低于T1(154 mm)、T2(137 mm)和T3(114 mm)处理。即在154~231 mm 降水处理下,降水量减小会显著提升土壤细菌群落的多样性(Shannon 指数)和丰富度(ACE 指数);当降水量低于154 mm 时,细菌群落多样性和丰富度指数未发生显著变化。
真菌群落的丰富度指数(Chao1 指数和ACE 指数)同样随着降水变化产生了显著性差异。即,当降水量介于154~231 mm 时,真菌丰富度指数随着降水减少呈显著升高趋势,超出这一降水范围后真菌丰富度指数则不会产生显著性差异;真菌多样性指数(Shannon 指数和Simpson 指数)并没有因为降水变化产生显著性差异。
2.4 不同降水梯度对荒漠草原土壤细菌及真菌群落β 多样性的影响
非度量多维尺度分析结果表明(图3),T0、T1、T2和T3处理下的土壤细菌和真菌群落在空间上彼此分离,各样本重复的组间距离较大,表明土壤细菌、真菌群落组成的相似性差异较大。采用ANOSIM 检验,进行999 次蒙特卡洛随机置换后发现,不同降水量分布的监测区内土壤细菌群落(r=0.37,P=0.001)和真菌群落(r=0.47,P=0.001)β 多样性差异显著。

图3 不同降水梯度下荒漠草原土壤细菌(a)和真菌(b)群落的非度量多维尺度分析Fig. 3 Non-metric multidimensional scaling analysis(NMDS) of bacteria(a) and fungi (b) in desert grasslands under different precipitation gradients
2.5 土壤环境因子对荒漠草原土壤微生物群落的影响
冗余分析结果表明(图4),环境因子显著影响细菌(P=0.002)和真菌群落(P=0.001)物种分布。在不同自然降水梯度下,TN(P=0.030)和AP(P=0.002)是驱动荒漠草原土壤细菌群落变化的主要环境因子,而驱动土壤真菌群落变化的环境因子则为OM 和TP。土壤细菌的丰富度指数(bacteria_Chao1 指数)与pH 呈正相关,与降水量、AP、TP 和TN 呈负相关;土壤细菌的多样性指数(bacteria_Shannon 指数)与pH 呈正相关,与降水量和AP呈负相关;细菌群落组成(bacteria_NMDS1)与TN 和TP 呈正相关,与降水量、AP 指数呈负相关。降水(R2=0.19,P=0.080)、TN(R2=0.29,P=0.030)、AP(R2=0.42,P=0.002)、AK(R2=0.04,P=0.420)、TP(R2=0.06,P=0.350)、pH(R2=0.07,P=0.310)、OM(R2=0.04,P=0.470)和植被生物量(R2=0.01,P=0.816)对细菌群落变异的解释率分别为13.40%、19.70%、35.70%、4.40%、1.20%、1.70%、0.80%和0.36%。环境因子对细菌群落变异的总解释率为77.16%(图4a)。

图4 不同降水梯度下荒漠草原土壤细菌群落(a)及真菌群落(b)与环境因子间的RDA 分析Fig. 4 Redundancy analysis (RDA) based on soil bacterial community (a), fungal community (b) and environmental factors under different precipitation gradients in desert grasslands
土壤真菌的丰富度指数(Chao1 指数)与pH、AP、OM 和AN 呈正相关,与降水量、植被多样性指数(plant_Shannon 指数)和AK 呈负相关;土壤真菌的多样性指数(fungi_Shannon 指数)与OM 和AN 呈正相关,与降水量呈负相关;真菌群落组成(fungi_NMDS1)与真菌多样性指数(fungi_Shannon 指数)、真菌丰富度指数(fungi_Chao1指数)、OM 和AN 呈正相关,与降水量呈负相关。降水(R2=0.44,P=0.09)、AK(R2=0.37,P=0.14)、OM(R2=0.46,P=0.03)、TP(R2=0.60,P=0.03)、AP(R2=0.05,P=0.34)、AN(R2=0.04,P=0.42)、pH(R2=0.03,P=0.72)和植被多样性(R2=0.01,P=0.81)对真菌群落变异的解释率分别为10.80%、9.90%、13.60%、14.20%、3.20%、1.60%、0.40%和0.20%。环境因子对真菌群落变异的总解释率为53.94%(图4b)。
为进一步揭示荒漠草原区降水量变化对土壤细菌和真菌的直接或间接影响,本研究基于RDA 分析结果以及与这些微生物群落有关的其他驱动因素(年均降水量、植物群落和土壤因子)之间的关系建立了结构方程模型(structural equation modeling, SEM)。SEM 结果显示(图5a),在以细菌群落为主的荒漠草原生态系统中,模型对植被群落生物量、植被多样性、土壤养分、细菌群落丰富度和群落组成的解释率分别为11%、2%、57%、46%和54%。荒漠草原区自然降水变化通过3 种途径对土壤细菌群落产生显著效应:1)直接对细菌群落丰富度产生极显著负效应;2)通过改变土壤养分间接地对细菌群落丰富度和群落结构产生显著正效应;3)通过影响植被群落生物量改变土壤养分,间接影响土壤细菌群落丰富度和群落结构。在真菌群落参与的荒漠草原土壤生态系统中(图5b),模型对植被群落生物量、植被多样性、土壤养分、细菌群落丰富度和群落组成的解释率分别为12%、2%、57%、13%和64%。降水通过2 种途径对真菌群落产生显著效应:1)通过改变土壤养分间接地对真菌群落结构产生极显著正效应; 2)通过影响植被群落生物量改变土壤养分,进而间接地对土壤真菌群落结构产生极显著正效应。

图5 荒漠草原生态系统各组分间的结构方程模型Fig.5 Structural equation model between biological and abiotic components of desert steppe ecosystem
3 讨论
3.1 降水量变化对荒漠草原区土壤细菌、真菌群落组成及相对丰度的影响
研究发现降水减少在一定程度上提高了土壤细菌群落和真菌群落的总OTUs,这表明干旱对土壤细菌和真菌群落丰富度会产生积极影响。此外,研究发现在不同降水梯度下,细菌总OTUs 均为真菌总OTUs 的2~3 倍,这与Chen 等[31]的研究结果类似。他们认为相较于真菌,土壤细菌的扩散作用更强,因而在荒漠草原生态系统中占据主要地位,是具有较高OTUs 的主要原因。
Na 等[32]和Wu 等[33]的研究结果表明,降水变化对优势细菌,如变形菌门、放线菌门、酸杆菌门和绿弯菌门的相对丰度影响不显著。然而,本研究却发现优势细菌中变形菌门、放线菌门、酸杆菌门和绿弯菌门在不同降水梯度下相对丰度差异显著。分析认为,不同优势细菌门类对降水变化的响应不一,主要是由于不同微生物类群具有不同的生活策略[34]。如,变形菌门作为典型的富养型细菌类群[35],环境资源越丰富,越有利于其生存和发展;而酸杆菌门作为寡营养类群,其丰度一般会随土壤养分的下降而升高[34]。
本试验中,土壤变形菌门相对丰度随降水减少呈显著降低趋势,在较高降水量的T0处理下更为富集;放线菌门、酸杆菌门和绿弯菌门的相对丰度总体上随着降水减少呈显著升高趋势,在降水量最低的T3处理下更为富集。研究认为,环境降水正常时,细菌类群与植被竞争荒漠土壤中有限的养分资源。由于变形菌门类群属于革兰氏阴性细菌,适宜的环境降水会保证土壤水分保持在正常状态,对革兰氏阴性细菌具有正向影响[36]。而当环境降水减少时,较低的土壤水分很可能会降低植物的蒸腾速率,从而降低元素的吸收,微生物对元素吸附能力的减弱也伴随着植物输入减少[37]。此外,干旱也会降低微生物活性[12],使得富养型细菌类群(如变形菌门)难以生存和发展。因此,土壤变形菌门细菌的相对丰度随着降水减少呈现出显著降低趋势。而对干旱和贫瘠环境条件具有较强抵抗力的寡营养类群细菌和耐旱型微生物类群(如酸杆菌门和放线菌门)能够超过其他微生物类群(如变形菌门)[38],故其相对丰度随降水减少呈显著升高趋势。土壤真菌各优势类群相对丰度并未随环境降水减少而产生显著性差异,这表明真菌群落整体上对降水减少具有一定的抵抗力;主要是因为真菌可以通过菌丝结构从缺水的土壤孔隙中获取足够的水分和养分、维系其正常的生命活动[14]。
3.2 降水变化影响下土壤细菌群落和真菌群落α 多样性差异
诸多研究表明,在许多生态系统中,细菌群落多样性普遍高于真菌多样性[31],本研究结果与之一致。这是因为许多土壤细菌类群具有抗旱性,随干旱程度加剧,细菌核糖体合成加快,能够在条件变得更有利于营养获取时占据优势地位[14]。Maestre 等[39]对除南极洲以外所有大陆的80 个旱地进行干旱效应评估,发现干旱程度加剧会降低土壤细菌和真菌的丰度和α 多样性。但本研究的结果与之相反,尽管在宁夏荒漠草原土壤上细菌群落和真菌群落丰度整体随降水减少呈上升趋势,α 多样性指数随干旱程度增加也呈显著上升趋势,但土壤真菌α 多样性指数对降水变化的响应不如细菌群落敏感。从微生物群落组成来看,基于门水平上相对丰度较高的前10 门细菌类群中,放线菌门、酸杆菌门、绿弯菌门、厚壁菌门、蓝菌门、疣微菌门以及其他未检出细菌类群的相对丰度整体上均表现为随降水量减小呈上升趋势,这可能是导致细菌丰度随降水减少而显著上升的主要原因。而真菌群落相对丰度较高的前10 门类群中,担子菌门和壶菌门等优势菌门以及未检出真菌类群的相对丰度整体上均表现为随降水量减小呈上升趋势。分析认为,这是导致真菌群落丰度随降水减少而显著上升的主要原因。徐鹏等[40]的研究也表明,不同真菌物种对干旱响应的差异性可能促使真菌群落组成变化,而真菌群落物种多样性相对恒定。
从微生物的生活策略而言,微生物对水分的适应状况取决于微生物本身的生理结构(如细胞壁)及特性[41]。在本研究中,真菌与细菌多样性指数对降水变化表现出不同的响应模式。相较于土壤细菌的单细胞结构而言,真菌的丝状结构能够重新分配利用水分[40],这使得真菌不易受水分含量变化的限制[42]。因此,真菌对干旱的适应能力较强[43],从而使真菌多样性对降水变化的敏感性不如细菌群落。细菌和真菌群落对水分变化的不同响应,也说明二者具有不同的生理特性。
3.3 降水变化下驱动荒漠草原土壤微生物群落变化的因子
Wang 等[44]认为,降水变化影响下植被多样性和生产力的变化是驱动土壤微生物变化的主要因素,本研究与其恰好相反。在本研究中,自然降水梯度作用下植物群落并未对土壤微生物群落产生明显驱动作用,这可能是因为宁夏中、北部荒漠草原植被群落以甘草、白草、短花针茅和牛枝子等沙、旱生植物为主[21],它们对外界环境变化具有较强的抵抗能力[45],植被群落多样性、生物量和群落组成特征较为稳定。
团队前期研究结果表明降水变化(114~231 mm)对荒漠草原土壤pH、有机质、全氮、碱解氮、总磷、速效磷和速效钾均产生了显著影响[21]。本试验结果表明土壤总氮和速效磷是驱动荒漠草原土壤细菌群落变化的主要环境因子,而驱动土壤真菌群落变化的环境因子则为有机质和总磷。诸多研究表明,土壤养分作为微生物的主要能源物质,能够影响微生物群落结构和群落组成[46-47]。随着自然降水量减少,植被对土壤养分元素的吸收减缓,使得土壤中碳、氮、磷等元素积累[48],这对微生物群落塑造产生了积极影响[47]。细菌和真菌作为土壤中主要的微生物类群,它们在形态特征、生长速率和底物利用方面存在很大差异[31],因此二者在资源利用策略和环境敏感性方面的反应不尽相同[49]。本研究中,土壤磷素是调节土壤微生物多样性的关键驱动因素,这一结果可能是由于所研究的荒漠草原具有很强的磷限制[12];此外,研究发现土壤细菌群落还受氮素驱动,真菌群落还受有机质驱动。综合分析认为,由于细菌与真菌具有不同的新陈代谢和生活策略;细菌对氮素的营养需求高于真菌,而真菌对碳的需求高于细菌[16,43],两者对不同土壤养分元素的需求差异,使得土壤细菌和真菌群落驱动因子存在差别。此外,作用于细菌群落和真菌群落的直接和间接生态因素存在差异;细菌群落和真菌群落同时受降水、植被和土壤养分调控,降水可以直接作用于土壤细菌群落;但无法直接作用于真菌群落,必须通过植被和土壤养分才能实现对真菌群落的影响。这是因为细菌的单细胞体形式无法跨越土壤中充满空气的孔隙,因此它高度依赖水进行移动和基质扩散[50];而真菌菌丝能够穿过充满空气的土壤孔隙获取养分和水分[51],这使得真菌群落对降水变化的抵抗性更高。然而,降水变化是如何通过影响植物、土壤养分间相互作用,影响土壤细菌和真菌群落的最终机制仍不十分清楚。因此,降水变化与土壤微生物群落间的作用关系及驱动机制还有待于进一步深入研究。
4 结论
荒漠草原土壤微生物群落受降水(114~231 mm)变化影响显著,降水减少对土壤细菌群落和真菌群落的丰富度均有促进作用,但真菌群落多样性指数对降水变化的敏感性低于细菌群落。在荒漠草原生态系统中,植被群落和土壤因子共同参与解释了土壤细菌群落和真菌群落对降水变化的响应过程,其中土壤因子的调控占主导作用,驱动细菌群落和真菌群落变化的环境因子也存在差异。荒漠草原生态系统土壤细菌和真菌群落应对降水量变化时的不同适应策略有助于预测未来降水变化趋势。
猜你喜欢
小哥白尼(趣味科学)(2022年5期)2022-08-15
中国石化(2021年8期)2021-11-05
中国比较医学杂志(2020年4期)2020-05-26
绿色中国(2019年14期)2019-11-26
启蒙(3-7岁)(2019年8期)2019-09-10
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
山东水利(2018年7期)2018-08-17
江西农业(2018年23期)2018-02-11
