
基于GWAS和WGCNA分析挖掘玉米花期相关候选基因
2023-11-15 12:09张占琴陈树宾丁永福桑志勤李卫华
作物学报 2023年12期
钱 甫 张占琴 陈树宾 丁永福 桑志勤,* 李卫华,*
基于GWAS和WGCNA分析挖掘玉米花期相关候选基因
钱 甫1张占琴2陈树宾2丁永福2桑志勤2,*李卫华1,*
1石河子大学农学院/ 绿洲生态农业兵团重点实验室, 新疆石河子 832000;2新疆农垦科学院, 新疆石河子 832000
花期是玉米重要性状之一, 解析玉米花期的遗传基础, 挖掘玉米花期关键基因, 对于选育广适玉米品种具有重要意义。在580份玉米自交系构成的自然群体中, 3年种植测定散粉期、吐丝期和散粉吐丝间隔期等3个花期性状, 利用分布全基因组的31,826个SNPs (single nucleotide polymorphisms)标记进行全基因组关联分析(genome wide association study, GWAS), 结合自交系B73的14个不同发育阶段的转录组数据进行权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA), 挑选与玉米开花相关的组织特异性模块和关键基因。GWAS在多环境(2个环境以上)下共定位标记14个, 挖掘到潜在候选基因10个, WGCNA挖掘到花期潜在候选基因17个, 2种方法共同挖掘到候选基因3个。编码一个MADS-box转录因子19,编码NAC转录因子133,编码MADS-box转录因子8, 这些基因主要参与调节花序生长发育。研究结果为解析玉米花期遗传基础及分子机制提供参考。
玉米; 花期性状; GWAS; WGCNA; 候选基因
玉米(L.)是饲料和生物能源的主要来源, 也是全球主要的粮食作物之一。在我国, 玉米是种植面积最大的作物, 也是对环境适应性最强的作物之一[1]。开花是玉米生命周期的关键组成部分, 成花诱导标志着其从营养阶段到生殖阶段的过渡, 开花时间可以反映玉米对环境的适应性, 适宜的开花时间对于玉米在特定环境中的生长发育至关重要[2]。玉米花期与品种的区域性和季节适应性密切相关, 影响穗的结实率[3], 因此花期性状不仅是评价品种熟性的重要指标, 也是决定玉米产量的重要性状之一。玉米花期性状是多基因控制的数量性状, 开花时间差异不是由少数具有较大效应的基因引起的, 而是由对性状的影响很小的许多数量性状位点的累积效应引起的[4]。
花期性状已在多种植物中得到广泛研究, 尤其是在拟南芥和水稻等作物中[5]。在拟南芥中已经鉴定出数百个控制开花时间的基因, 并且构建了一个相对完整的开花时间基因调控网络[6]。随着玉米基因组序列测序完成, 玉米开花期的遗传研究也取得显著进展。Dong等利用40多个玉米开花时间候选基因构建了开花时间遗传控制的基因调控网络, 并以、、和等4个开花基因为基础建立花期动态基因调控网络(dynamic gene network, DGN)模型预测玉米开花时间, 将玉米开花诱导调控网络分为光周期途径、自主途径、赤霉素途径和年龄途径等四大调控途径[7]。虽然前人已经构建了玉米开花时间基因调控网络, 并且一些研究者通过GWAS等方法挖掘花期性状候选基因, 如李真等以248份玉米自交系材料进行关联分析挖掘到花期候选基因编码early flowering 4蛋白, 参与光周期和昼夜节律的调节,编码GID1蛋白, 参与植物激素信号转导过程和赤霉素代谢[8]; Zhang等利用310份玉米自交系挖掘到16个开花性状候选基因主要涉及自主、光周期、花粉萌发等途径[3]; Yuan等[9]在干旱和热胁迫条件下挖掘到3个已报道开花基因、和, 也发现新的候选基因编码光敏色素A相关F-box蛋白。但相比于拟南芥等作物, 仍有许多调控玉米开花时间的基因尚未得到发现及研究, 并且在玉米中只有少量与开花时间相关的基因被报道, 如[10]、[11]、[12]、[13]、[14]、[15]、[16]、[17]、[18]和[19]等。
随着分子标记技术和测序技术的飞速发展, 多种作物已基本完成全基因组测序, 基于SNPs标记的全基因组关联分析已广泛应用于解析作物表型性状变异遗传机制[20]。海量的测序数据需要分析, 权重基因共表达网络分析(WGCNA)作为一种系统生物学方法, 已广泛应用于多样本的生物学问题研究中[21]。WGCNA利用生命活动互相关联的特点, 结合高通量测序技术获得基因表达量将具有相似表达模式的基因划分到同一模块, 研究共表达模块和目标性状之间的生物学相关性, 探索基因网络与性状之间的关联关系, 挖掘与性状高度关联的关键基因[22]。
虽然玉米花期性状的遗传机制取得相应研究, 但仍需在前人研究基础上继续挖掘与解析花期基因来完善玉米开花时间基因调控网络。因此, 利用不同的群体材料和测序技术, 在不同环境下采用多种方法进行遗传解析, 得到相同或不同的研究结果对解析玉米花期性状的遗传基础有一定的参考意义。本研究利用580份玉米自交系构成的自然群体为试验材料, 利用基于靶向测序分型(genotyping by target sequencing, GBTS)技术开发的Maize 40K育种芯片进行全基因组测序, 对玉米花期性状进行全基因组关联分析。同时结合玉米自交系B73的14份不同发育阶段的转录组数据进行权重基因共表达网络分析, 挖掘与玉米花期相关的SNPs 位点和候选基因, 为玉米分子标记辅助育种提供理论支持。
1 材料与方法
1.1 试验材料和试验设计
试验材料来源为自育自交系及引进加拿大早熟改良群、BSSS系、NSS系、黄淮海种质、欧洲KWS系列和先锋系列等580份自交系构成的自然群体, 由新疆农垦科学院作物研究所提供。于2019年、2020年和2021年在新疆农垦科学院作物研究所玉米育种试验田种植(44.31°N, 85.99°E), 田间设计采用拉丁方, 种植密度105,000株hm–2, 2次重复, 单行区, 行长4.50 m, 行距0.55 m, 田间管理与大田生产相同。
玉米不同发育阶段的转录组数据从MaizeGDB (https://www.maizegdb.org/)获得, 转录组数据来源于玉米B73材料的叶片、顶端分生组织、花丝、花药、果穗、根等组织[23]。
1.2 表型调查
散粉期(days to anthesis, DTA)记录从出苗到50%以上的植株雄穗散粉的天数; 吐丝期(days to silking, DTS)记录从出苗到50%以上的植株雌穗吐丝的天数; 散粉吐丝间隔期(anthesis silking interval, ASI)植株散粉和吐丝间隔的天数。
1.3 表型数据处理
利用R软件处理表型数据, 进行描述性统计分析、方差分析和相关性分析, 并绘制相应的图形。广义遗传力按照Knapp等[24]提出的公式2=g2/ (g2+gy2/+e2/)计算, 其中g2为遗传方差,gy2为基因型与年份互作的方差,e2为误差方差,为年份数,为重复数。为了消除环境(年份)变异对表型值的影响, 利用R包lme4构建混合线性模型来估计花期性状的最佳线性无偏估计值(best linear unbiased estimate, BLUE)值, 用于后续全基因组关联分析。
1.4 基因型数据分析
采取CTAB (cetyltriethylammnonium bromide)法提取各个材料叶片组织的DNA, 检测DNA提取质量。利用基于GBTS技术开发的Maize 40K育种芯片进行全基因组测序分析(该部分工作由石家庄博瑞迪生物技术有限公司完成)。利用PLINK1.9软件对基因型数据进行质控, SNPs的筛选标准为最小的等位基因频率MAF (minor allele frequency)>0.05, 基因缺失率<0.05, 筛选出高质量的SNPs标记31,826个。
1.5 全基因组关联分析
使用R软件GAPIT包的FarmCPU (fixed and random model circulating probability unification)模型进行全基因组关联分析, 以PCA (principal components analysis)和K (亲缘关系)矩阵作为关联分析的协变量来降低假阳性。FarmCPU模型适用于大数据分析, 计算速度快, 统计效力高[25]。为了控制假阳性的概率, 采用Bonferroni校正(=0.05/,为SNPs标记数)对阈值进行调整。以=1.57×10–6作为显著相关的阈值水平线选择显著SNPs标记。
1.6 候选基因筛选
根据与表型性状显著关联的SNPs标记在玉米B73参考基因组(ZmB73_RefGen_v4)中的物理位置, 以及2=0.1时染色体LD (linkage disequilibrium)的平均衰减距离, 筛选显著SNPs标记上下游LD范围内的候选基因。利用MaizeGDB及NCBI (https:// www.ncbi.nlm.nih.gov/)对候选基因进行功能注释和预测。
1.7 权重基因共表达网络分析
利用R软件WGCNA 包构建权重基因共表达网络[26], 基因的表达谱矩阵来自所有样本的基因表达量。考虑到在各个组织中均低表达的基因可能不具有生物学意义, 过滤掉最高表达量FPKM (fragments per kilobase of exon model per million mapped frag-ments)值小于5的基因[21]。为了使网络符合无尺度网络分布, 利用 WGCNA 提供的pickSoftThreshold函数计算软阈值(设置powers = c(c(1:10), seq (from= 12, to=20, by=2))), 选择拟合曲线第一次接近0.85时的阈值参数值构建基因共表达网络, 利用动态剪切法识别共表达模块。计算每个模块的特征向量值ME (module eigengene)并对ME聚类, 选择相似性在75%以上模块进行聚类合并(设置deepSplit=2、minModuleSize=30、MEDissThres= 0.25)[27]。表达模式相同的基因可能具有相似的生物学功能, 这些基因可划分到同一模块中, 这样的模块具备高度的生物学意义。为了深入研究与组织高度相关联的模块, 将不同组织作为性状, 创建表型矩阵, 计算ME和不同组织之间的相关系数, 相关系数越大, 模块与组织间关联度越高, 而相关系数越小, 关联度越低。为挖掘具有生物学意义模块, 相关系数高于0.65被定义为组织特异性模块[28]。将组织特异性模块中的基因上传到在线网站(https://www.omicshare.com/ tools/Home/Soft)进行GO (gene ontology)富集分析, 利用开花调控相关的一些关键词例如开花(flower)、光(light)等, 在组织特异性模块中查找参与调控开花的模块与基因。查找模块中文献已报道过的开花基因和模块中高连通性基因(连通性排名前10%)作为模块的枢纽基因[21], 利用这些基因进行互作网络的构建, Cytoscape软件对局部调控网络进行可视化。
2 结果与分析
2.1 花期性状表型数据分析
花期性状在不同年份下表现出广泛的连续变异, 其中散粉吐丝间隔期表型变异最大, 变异系数为35.88%~61.87%, 其他2个花期性状变异系数在7.86%~9.54%之间。花期性状的偏度和峰度的绝对值均小于1, 表明不同年份下性状均服从正态分布。除散粉吐丝间隔期性状外, 散粉期和吐丝期性状均表现出较高的广义遗传力, 分别为96.18%和95.54% (表1), 说明花期性状遗传主要受基因的控制。
方差分析表明(表2), 3个花期性状在基因型效应、环境(年份)效应、基因型与环境互作效应方面均表现出极显著水平, 说明花期性状受基因型、环境和基因型与环境互作的影响。
相关性分析(图1)可知, 在不同年份, 散粉期和吐丝期表现出极显著正相关, 表明这2个性状的生长发育存在紧密得协同促进作用。散粉吐丝间隔期主要与吐丝期存在显著正相关, 说明散粉吐丝间隔期主要与玉米吐丝时间有关, 相比于散粉期和吐丝期性状, 散粉吐丝间隔期性状更易受到环境影响。
2.2 全基因组关联分析
使用FarmCPU模型以PCA和K矩阵作为关联分析的协变量进行全基因组关联分析, 由曼哈顿图和QQ图(图2和附图1)可知, 假阳性得到较好的控制。
经过Bonferroni校正, 3个花期性状在4个环境下(3个年份和BLUE值)共检测到95个显著关联SNPs标记(附表1)。为保证关联结果的准确性, 选择2个环境以上共同定位到的SNPs标记进行后续分析。散粉期、吐丝期和散粉吐丝间隔期等3个花期性状的共定位标记分别为9、8和1个, 除10号染色体外, 其他染色体均有分布(表3)。除散粉吐丝间隔期性状外, 其他2个花期性状之间也有共同的SNPs标记(图3), 不同性状之间共定位标记说明该标记可能存在一因多效性, 雄穗和雌穗的部分生长发育进程可能受到相同的基因控制, 有相似的遗传基础。
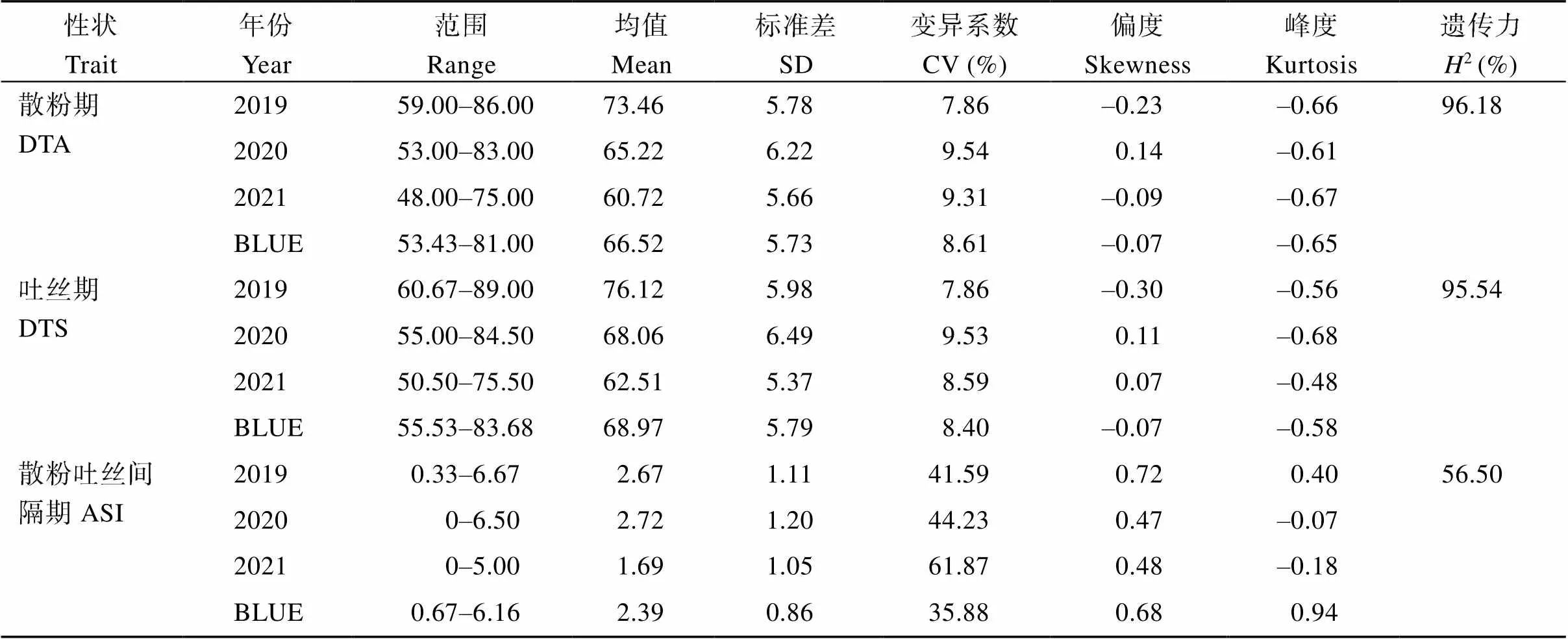
表1 不同年份下花期性状描述统计分析
DTA: 散粉期; DTS: 散粉期; ASI: 散粉吐丝间隔期。
DTA: days to anthesis; DTS: days to silking; ASI: anthesis silking interval.

表2 花期性状方差分析
***表示在0.001概率水平差异显著。缩写同表1。
***indicates significant difference at the 0.001 probability level. Abbreviations are the same as those given in Table 1.
**和***分别表示在0.01和0.001概率水平差异显著。缩写同表1。
**and***indicate significant in the 0.01 and 0.001 probability levels, respectively. Abbreviations are the same as those given in Table 1.
缩写同表1。Abbreviations are the same as those given in Table 1.
2.3 等位基因变异效应分析
对花期性状不同环境下及多个性状之间共同检测到的SNPs标记进行等位基因变异分析(检验)发现, 所有共定位SNPs标记不同等位基因型的表型性状至少在3个环境下均达到显著水平(图4和附图2), 表明这些SNPs标记对花期性状表型变异具有一定的影响。其中5_176849292、8_159342177、3_135182164和8_130284569 是多个性状之间共定位标记, 在4个环境下, 5_176849292标记GG基因型比AA基因型散粉平均缩短3.62~4.28 d, 吐丝平均缩短3.27~4.42 d; 8_159342177标记TT基因型比CC基因型散粉平均缩短2.33~2.92 d, 吐丝平均缩短2.46~3.18 d; 3_135182164标记AA基因型比TT基因型散粉平均缩短3.68~4.37 d, 吐丝平均缩短3.82~4.39 d; 8_130284569标记CC基因型比TT基因型散粉平均缩短4.04~5.08 d, 吐丝平均缩短3.47~5.09 d。本研究检测到4个一因多效位点, 即一个SNPs标记可以控制多个花期性状的表型。

表3 花期性状显著共定位SNPs标记
缩写同表1。Abbreviations are the same as those given in Table 1.
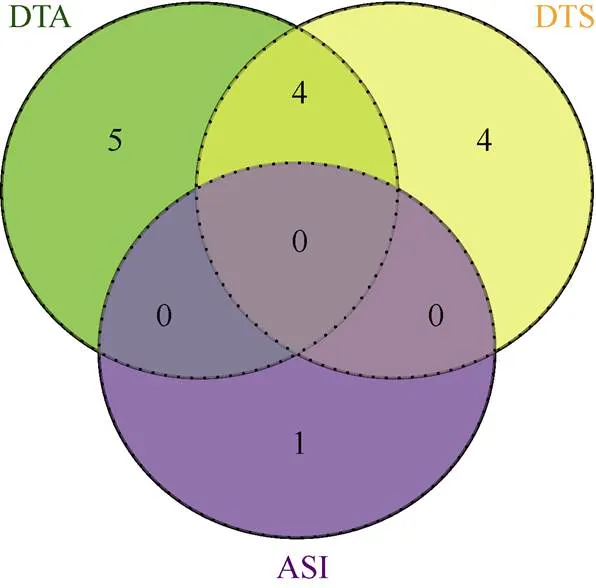
图3 花期性状显著共定位SNPs数量
缩写同表1。Abbreviations are the same as those given in Table 1.
2.4 权重基因共表达网络分析
根据FPKM值, 将玉米基因表达矩阵中表达量较低的基因进行过滤, 获得19,959个高表达基因。基因表达水平的样本聚类和性状关联结果表明每个组织的基因聚类树可以和组织很好地对应(图5-A)。根据软阈值计算结果(图5-B), 选择=7来构建网络, 采用动态剪切树法合并表达相似的模块, 共获得16个共表达模块, 用不同的颜色来表示每个模块(图5-C), Turquoise模块内基因最多, Grey模块是一组未分配到其他任何模块的基因。
16个模块中有10个与组织存在高度特异性(图5-D), 大部分组织都有与其高度相关的模块。10个组织特异性模块都富集到与开花相关的调控通路(图5-E), 查阅相关文献, Blue模块包含1个已报道开花基因[10], Brown模块包含5个已报道开花基因[12]、[17]、、[29]和[30], Green模块包含4个已报道开花基因[11]、[31]、[32]和.7[33], Purple模块包含3个已报道开花基因[31]、和[34]。Black模块与花丝紧密相关(=0.94,=5E–07), Grey60模块与V5过渡叶底部紧密相关(=0.77,=0.001), Royalblue模块与V18雄穗紧密相关(=0.77,=0.001), Lightcyan模块与V5第1伸长节间紧密相关(=0.88,=3E–05), Darkgrey模块与V3茎尖端分生组织紧密相关(=0.86,=7E–05)。花丝、雄穗、叶片等组织是与玉米开花密切相关的组织[21,35-36], 因此本研究重点关注这9个模块。利用模块中已报道的开花基因或高连通性基因作为模块中的核心基因构建基因互作网络, 筛选与花期相关候选基因。
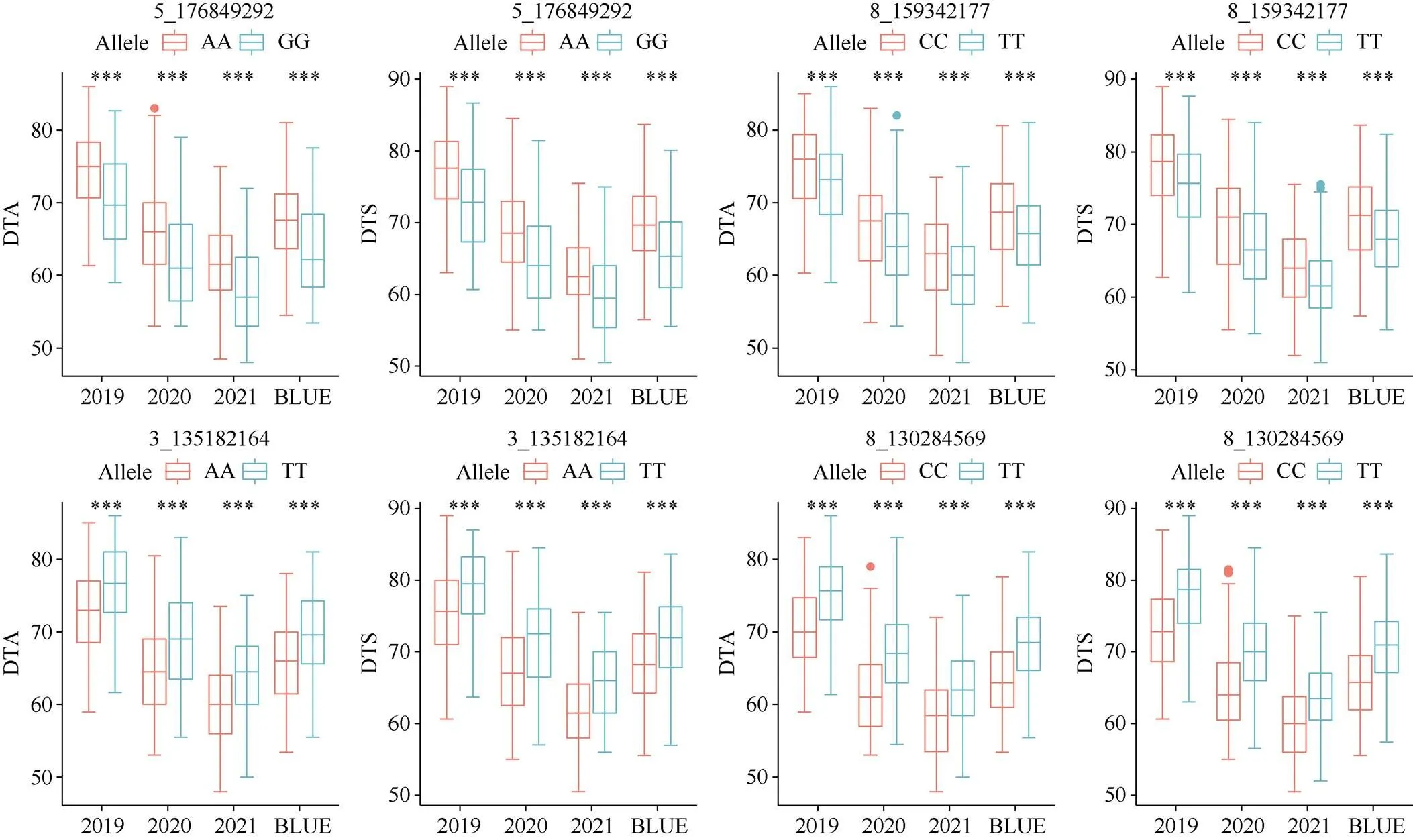
图4 花期性状共定位SNPs标记等位基因效应分析
***表示在0.001概率水平差异显著。缩写同表1。
***indicates significant difference in the 0.001 probability level. Abbreviations are the same as those given in Table 1.
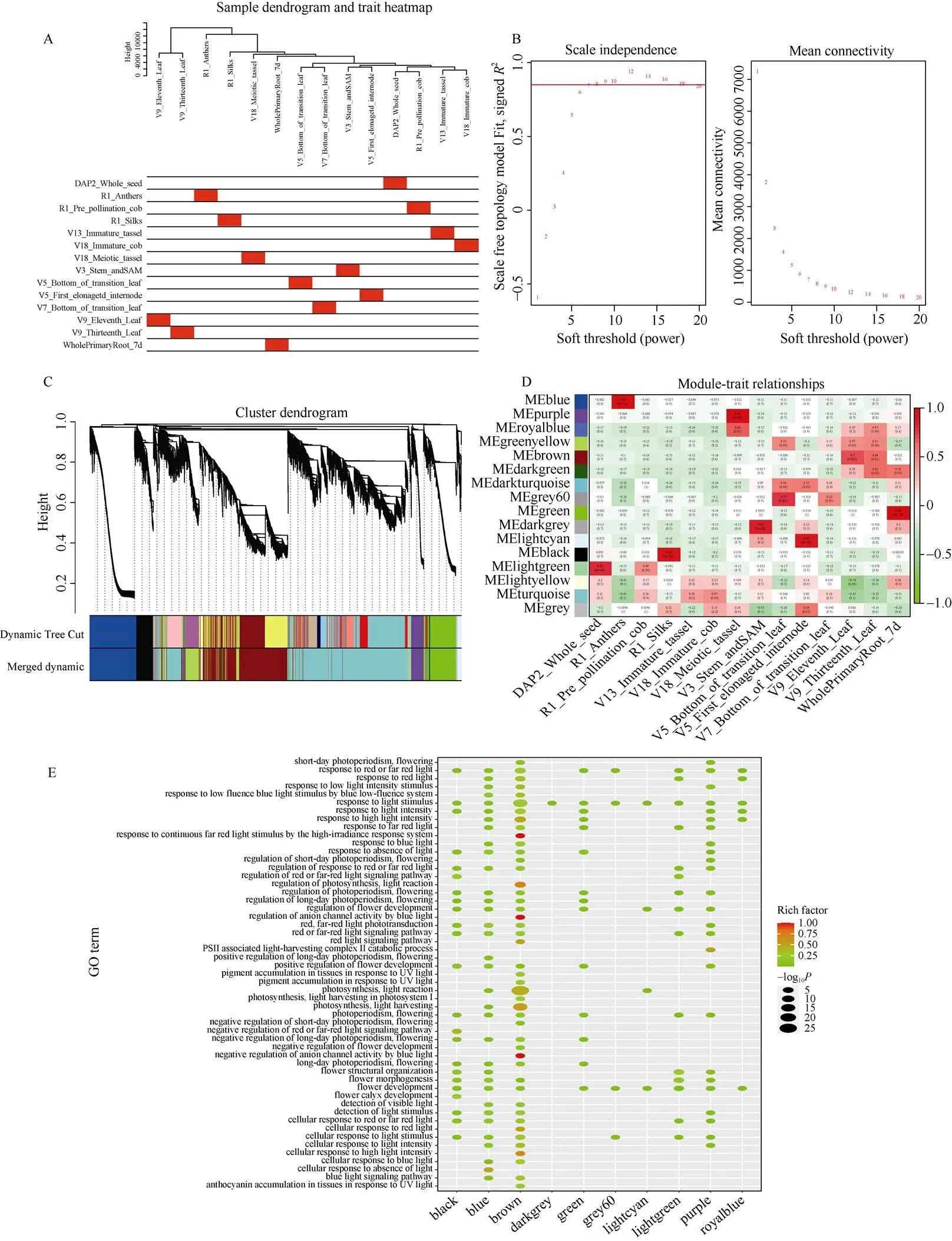
图5 基因共表达网络构建结果
A: 样本层次聚类树及组织对应; B: 软阈值确定; C: 基因聚类和模块构建; D: 性状与模块相关性分析(红色的格子代表性状与模块具有正相关性, 绿色的格子代表性状与模块具有负相关性, 图中括号内外的数值分别为值和相关系数); E: 开花富集通路。
A: the clustering dendrogram of samples and tissue correspondence; B: the determination of soft threshold; C: gene clustering and module construction; D: correlation between traits and modules (Red color of each box represents the positive correlation between module and trait. Green color of each box represents the negative relationships between module and trait. The values inside and outside the brackets in the figure are-values and correlation coefficients, respectively); E: enrichment pathway of flowering time.
2.5 候选基因分析
基于该群体LD衰减距离440 kb (2=0.1), 在B73_RefGen_v4参考基因组上显著共定位标记上下游440 kb范围内查找相关候选基因, 共检测到173个基因, 经功能注释筛选出10个可能与开花时间相关的候选基因(表4)。其中和已在玉米中证实与开花时间有关。编码WD重复蛋白RBAP1 (), Mascheretti等人发现/蛋白与玉米开花基因和直接相关, 调节玉米开花途径基因的表达,/突变品系与野生型相比开花延迟[37]。编码Corngrass1 (),通过其下游靶标转录因子参与开花的调控[38], 且过表达会延迟开花[39]。除已报道基因, 还筛选出8个具有不同功能的新的花期候选基因, 它们在拟南芥中的同源基因得到研究证实与开花时间有关, 但尚未在玉米中得到研究, 如编码PHD转录因子30, 拟南芥同源基因()编码调节开花时间的染色质重塑因子,通过特异性抑制基因的表达来参与开花时间的调节[40]。编码6-磷酸果糖-2-激酶/果糖-26-二磷酸酶, 拟南芥同源基因()编码双功能酶果糖-6-磷酸2-激酶/果糖-26-二磷酸酶,突变体在8/16 h的明暗循环中生长会延迟花卉的萌发[41]。
通过WGCNA分析, 利用玉米开花相关的组织特异性模块中已报道开花基因及模块内高连通的核心基因筛选与玉米花期相关的候选基因, Blue模块利用1个已报道开花基因筛选出2个花期候选基因, Brown模块利用5个已报道基因筛选出3个候选基因, Green模块利用4个已报道基因筛选出3个候选基因, Purple模块利用3个已报道基因筛选出1个候选基因, Black、Grey60、Lightcyan和Royalblue模块分别从模块内高连通基因中筛选除4、1、2和1个花期候选基因(图6), Darkgrey模块可能由于聚类基因较少, 所以并未筛选到与花期相关的候选基因。在9个组织特异性模块中筛选出17个花期相关候选基因(表5), 其在拟南芥中的同源基因均得到充分研究与开花时间有关, 如编码MYB相关转录因子96, MYB 转录因子在植物花的发育过程中发挥着重要的调控作用, 直接或间接地与某些外源或内源刺激发生相互作用, 从而影响植物开花时间[42]。的拟南芥同源基因编码 MYB 家族转录因子, 参与拟南芥的昼夜节律调节,的表达受光调节, 在长日照条件下过表达延迟开花[43]。编码甘露糖-1-磷酸鸟苷酸转移酶2, 其拟南芥同源基因()参与抗坏血酸生物合成, 抗坏血酸控制开花时间和衰老,突变体在长日照条件下提早开花, 短日照条件下延迟开花[44]。编码CO CO-LIKE TIMING OF CAB1结构域蛋白24 (), 许多含CCT结构域的蛋白质是参与开花植物光周期敏感性的转录因子[45], 玉米CCT结构域蛋白基因对玉米的光周期敏感性特别重要, 基因高表达导致玉米在长日照下开花延迟[19]。编码ALG2相互作用蛋白X, 其拟南芥同源基因()的突变具有多效性, 导致植物生长缓慢延迟开花[46]。编码Aux/IAA转录因子4 (), Aux/IAA蛋白是生长素信号转导的中枢调节剂, 参与控制植物发育的许多方面, 其拟南芥同源基因()在拟南芥开花时间调节中起作用,突变体在短日照条件下晚开花,可能通过负调节和基因的表达来抑制短日照条件下开花时间[47]。编码BAG家族分子伴侣调节因子4, BAG家族是一类在多种信号通路中发挥作用的多功能蛋白, 在植物生长发育过程中发挥重要作用, 其拟南芥同源基因()突变植株表现出提早开花和衰老[48]。编码肌动蛋白解聚因子4 (), 肌动蛋白解聚因子(ADF)是一种肌动蛋白结合蛋白, ADF家族在植物生长、发育和防御相关功能中起着关键作用[49], 其拟南芥同源基因()沉默会延迟开花[50]。
3 讨论
花期性状是玉米生长发育育周期中的重要农艺性状, 是育种工作中重点关注的主要性状, 涉及营养生长到生殖生长的不可逆转换, 在生态环境适应性及对于玉米的产量、质量、营养价值和抗性等多个方面都具有非常重要的影响[51], 解析玉米花期性状的遗传基础具有重要意义。
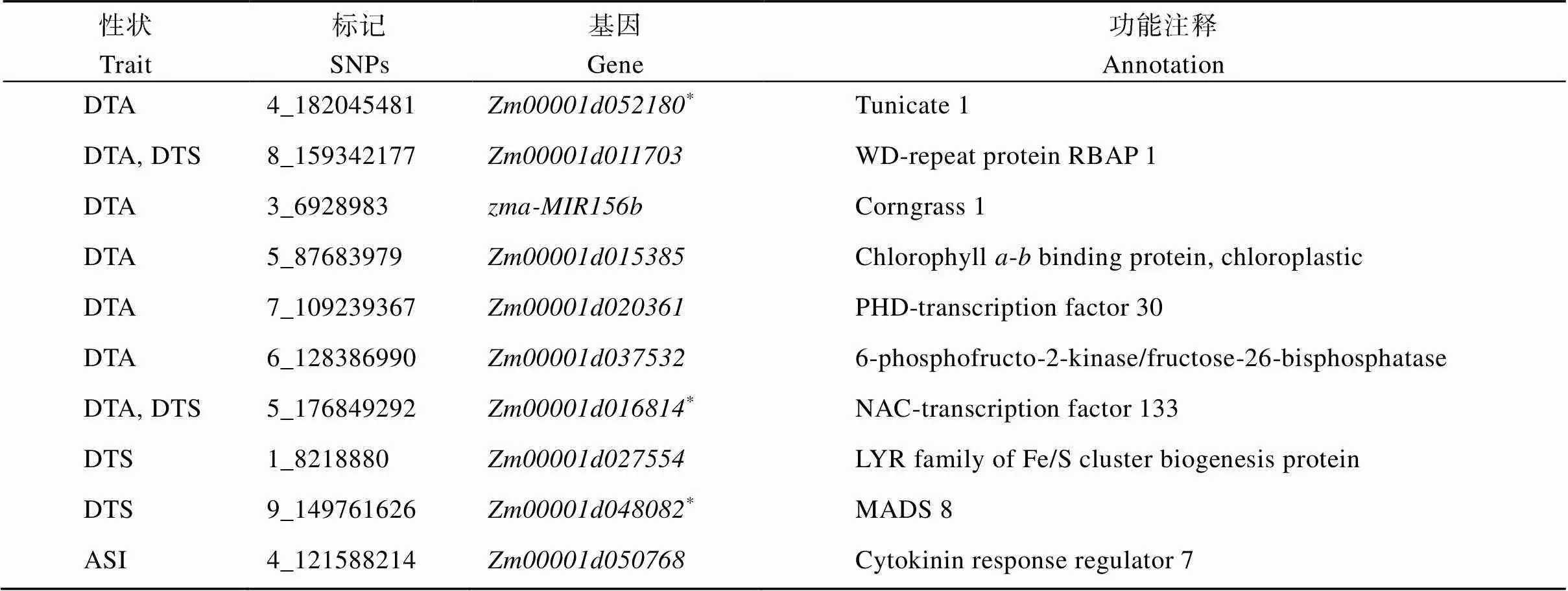
表4 GWAS候选基因
*表示该基因为GWAS和WGCNA共同检测到。缩写同表1。
*The gene co-mined for GWAS and WGCNA. Abbreviations are the same as those given in Table 1.
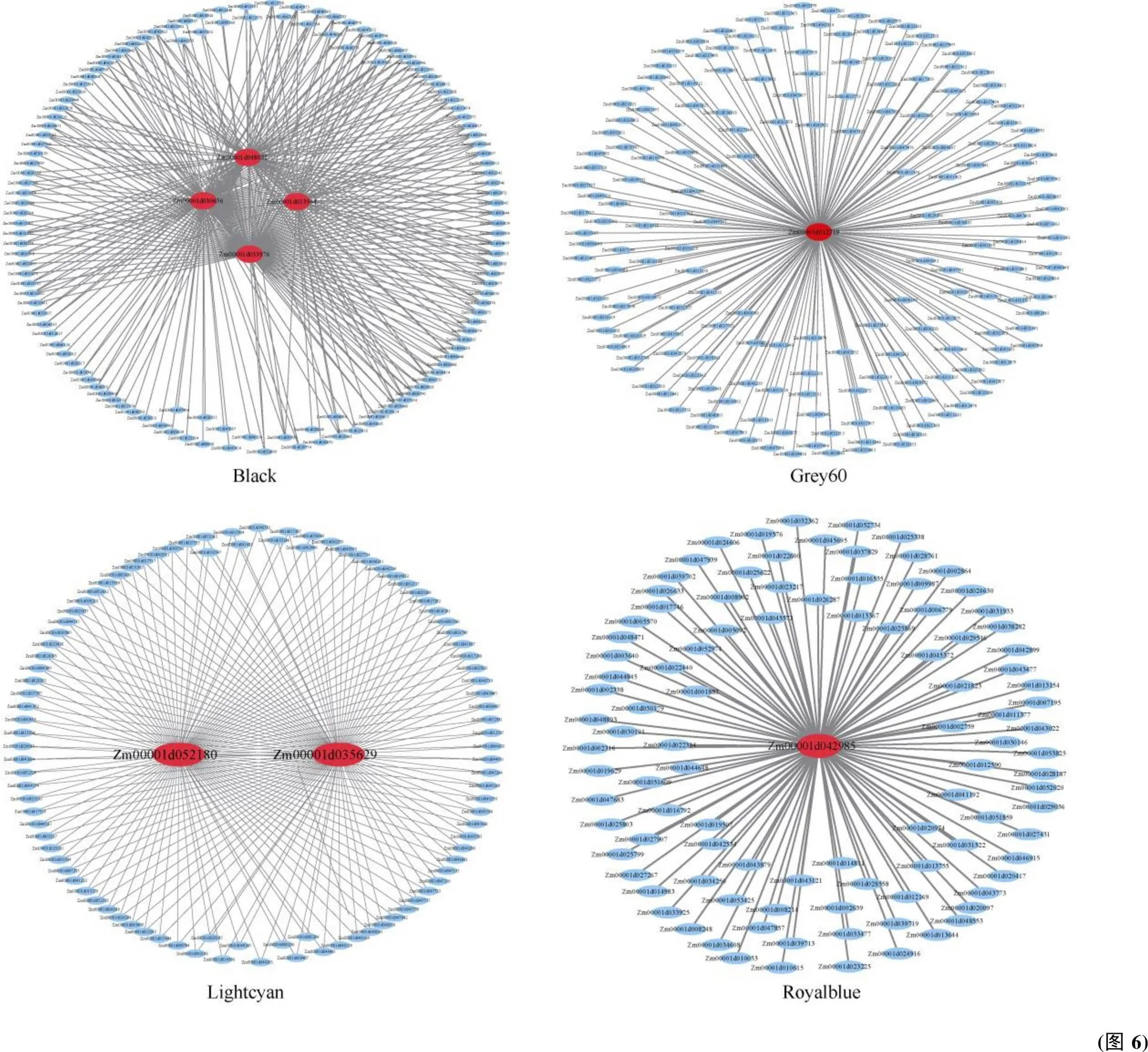
绿色节点是已报道开花基因, 红色节点是筛选出的开花候选基因。
The green node is the flowering time genes reported, the red nodes are flowering time candidate genes.
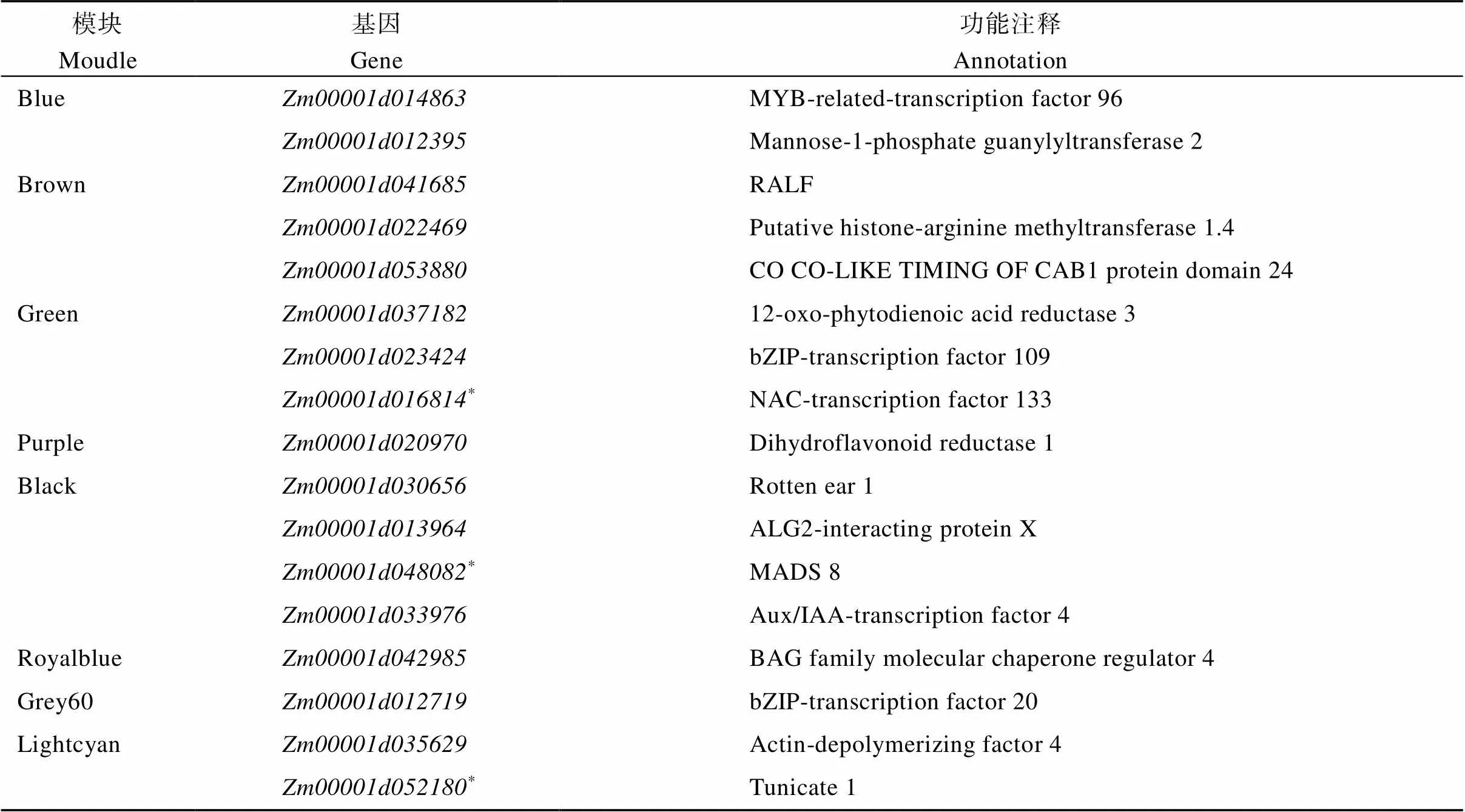
表5 WGCNA候选基因
*表示该基因为GWAS和WGCNA共同检测到。
*indicates the genes co-mined for GWAS and WGCNA.
3.1 花期性状共定位SNPs标记研究
目前已知玉米花期性状QTL (quantitative trait loci)热点区域主要为bin1.01、bin1.03、bin3.04、bin3.05、bin4.08、bin8.05、bin9.06、bin10.04等区段[9,52], 本研究检测到的部分共定位标记位于这些热点区域内, 如标记1_8218880与Wang等[53]、Khairallah等[54]检测到的QTL同位于bin1.01, 3_135182164与侯清桂等[55]、杨慧丽等[56]、Li等[36]检测到的QTL同位于bin3.05, 4_182045481与杨慧丽等[56]、袁亮等[57]、Shi等[58]检测的QTL同位于bin4.08, 6_63992603与李真等[8]、魏海忠等[59]、Zhang等[3]检测到的QTL同位于bin6.01, 6_128386990与李真等[8]、李凯等[60]、袁亮等[57]、Maldonado等[2]检测的QTL同位于bin6.05, 8_130284569与郭向阳等[61]、袁亮等[57]检测到QTL同位于bin8.05。一些标记未与前人定位结果重叠或接近, 如散粉期相关标记3_6928983及散粉吐丝间隔期相关标记4_ 121588214。与前人定位结果在相同或相近bin区段的SNPs标记, 在一定程度上说明了本研究定位结果的准确性[8], 而未与前人研究定位到相同bin区段的SNPs标记, 可能是新发现的有意义的功能位点或标记。本研究检测到的所有共定位SNPs标记对花期性状表型均有显著影响, 其中多个性状之间共同定位的SNPs能同时调控多个花期性状, 表明这些SNPs具有一因多效性, 可能与控制花期性状的多个基因或一个多效性基因连锁, 是潜在用于玉米花期性状遗传改良的重要遗传标记或位点。这些研究结果可以为筛选不同熟性的材料提供帮助, 为分子标记辅助育种技术培育早熟性品种提供理论支持。
花期性状在不同年份下均检测出较多的显著性SNPs标记, 但在多环境下的共定位标记较少, 一方面可能是花期性状主要是多基因控制的数量性状, 受环境影响较大, 不同年份下的环境条件不同可能导致控制性状的基因在不同条件下进行差异表达[62],多环境共检测到的基因是稳定表达的基因, 因此多环境下共定位标记相对较少。另一方面可能与本研究质控后的高质量SNPs标记(31,826个)数量相对较少, 密度较低有关。
3.2 玉米花期相关的调控途径
植物开花途径一直是研究的热点, 前人通过对开花诱导调控网络的研究, 将玉米开花途径分为光周期途径、自主途径、赤霉素途径和年龄途径[51]。不断深入研究玉米花期性状, 解析花期遗传基础和明确开花基因的分子作用机制可以完善开花调控网路, 准确完整的调控网络不仅加深人们对花发育机制的认知与理解, 还指导玉米育种和未来的研究方向和策略[7]。本研究筛选到的候选基因涉及光周期途径、自主途径、赤霉素途径和年龄途径, 如参与年龄途径调控,是年龄途径中关键的miRNA基因[7,51]。()可能参与赤霉素途径调控, 其拟南芥同源基因()可能通过负调节和基因的表达控制开花[44]。(), 与玉米开花基因直接相关, 调节玉米开花途径基因的表达,是玉米的自主开花途径的关键基因[52], 因此可能参与自主途径调控。的拟南芥同源基因是(),是拟南芥光周期途径中的关键基因[63];编码叶绿素结合蛋白, 其拟南芥同源基因()参与光系统I反应[64];()的拟南芥同源基因()的表达受光调节, 参与光周期昼夜节律途径[43];()是具有CCT结构域的ZmCCT家族的基因, ZmCCT家族是光周期途径的核心基因家族[65], 这些候选基因可能主要参与光周期途径。
3.3 GWAS和WGCNA共同挖掘花期相关候选基因
本研究利用GWAS和WGCNA从基因组学和转录组学进行玉米花期性状遗传解析, 虽然GWAS方法已被广泛用于揭示玉米表型变异的遗传基础[66-67],但GWAS也具有一定的局限性, 如开花、产量等许多复杂的农艺性状是一个与群体结构存在高度相关的性状, 利用GWAS鉴定目标性状相关的调控基因容易受到群体结构的影响[21], 并且GWAS对一些微效多基因控制的数量性状检测能力不足, 很难准确挖掘出微效作用位点或基因, 在检测的过程中也会引入假阳性或假阴性结果, 可能造成实验结果不准确[27]。WGCNA是分析基因共表达调控的有效方法, 能够特异地筛选与性状具有高度生物学意义的共表达模块以及挖掘关键基因, 能较好地解决GWAS对复杂性状解析能力不足的问题, 已被证明是一种高效的数据挖掘方法[68]。因此, 基因组和转录组的多组学结合, GWAS和WGCNA的联合分析是获取花期性状关键基因的有效途径, 是解析花期遗传基础的有效策略, 能互相弥补各自方法的缺陷与不足[66,69-70]。本研究利用GWAS 和WGCNA分别挖掘到10个和17个花期性状候选基因, 2种方法共同挖掘到、和这3个基因,()编码一个MADS-box转录因子()参与玉米雄穗花序发育,为拟南芥开花时间调节蛋白的同源基因, 抑制开花[71]。编码NAC转录因子133, NAC转录因子是植物特有的一类转录因子家族, 在植物花的发育过程中具有重要的调控作用, 可以调控开花时间[72], 如拟南芥NAC转录因子的过表达导致开花延迟[73],调控水稻株高和开花时间[74], 大豆过表达植株早开花[75]。编码MADS-box转录因子8, 参与控制花序分生组织, 调节花序生长,与在花序分生组织中的时空错误表达造成玉米穗不分枝[76]。这些基因有很大可能性参与调控玉米开花时间, 但仍需进一步进行基因功能分析加以验证。
4 结论
本研究利用GWAS和WGCNA两种方法共同挖掘玉米花期性状候选基因24个,、、-、、、、、、、,、、、、、、、、、、、、、。其中3个(、、)是2种方法共同检出, 从基因组和转录组2个方面为解析玉米开花遗传基础提供了参考依据。
附图和附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical- zuowxb.aspx。
[1] Liu M, Tan X L, Yang Y, Liu P, Zhang X X, Zhang Y C, Wang L, Hu Y, Ma L L, Li Z L, Zhang Y L, Zou C Y, Lin H J, Gao S B, Lee M, Lübberstedt T, Pan G T, Shen Y. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping., 2020, 18: 207–221.
[2] Maldonado C, Mora F, Bertagna F A B, Kuki M C, Scapim C A. SNP-and haplotype-based GWAS of flowering-related traits in maize with network-assisted gene prioritization., 2019, 9: 725.
[3] Zhang H Y, Gao S, Li B Y, Zhong H X, Zhang Z C, Luo B W. Genome-wide association analysis of maize flowering traits., 2020, 12: 43–46.
[4] Buckler E S, Holland J B, Bradbury P J, Acharya C B, Brown P J, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz J C, Goodman M M, Harjes C, Guill K, Kroon D E, Larsson S, Lepak N K, Li H H, Mitchell S E, Pressoir G, Peiffer J A, Rosas M O, Rocheford T R, Romay M C, Romero S, Salvo S, Sanchez Villeda H, Da Silva H S, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu J M, Zhang Z W, Kresovich S, Mcmullen M D. The genetic architecture of maize flowering time., 2009, 325: 714–718.
[5] Shrestha R, Gómez-Ariza J, Brambilla V, Fornara F. Molecular control of seasonal flowering in rice,and temperate cereals., 2014,114: 1445–1458.
[6] Li Y X, Li C H, Bradbury P J, Liu X L, Lu F, Romay C M, Glaubitz J C, Wu X, Peng B, Shi Y S, Song Y, C Zhang D F, Buckler E S, Zhang Z W, Li Y, Wang T Y. Identification of genetic variants associated with maize flowering time using an extremely large multi-genetic background population., 2016, 86: 391–402.
[7] Dong Z, Danilevskaya O, Abadie T, Messina C, Coles N, Cooper M. A gene regulatory network model for floral transition of the shoot apex in maize and its dynamic modeling., 2012, 7: e43450.
[8] 李真, 刘文童, 杨硕, 郭晋杰, 赵永锋, 黄亚群, 陈景堂, 祝丽英. 玉米花期性状的全基因组关联分析. 分子植物育种, 2020, 18: 37–45. Li Z, Liu W T, Yang S, Guo J J, Zhao Y F, Huang Y Q, Chen J T, Zhu L Y. Genome-wide association analysis of flowering time related traits in maize (L.)., 2020, 18: 37–45 (in Chinese with English abstract).
[9] Yuan Y B, Cairns J E, Babu R, Gowda M, Makumbi D, Magorokosho C, Zhang A, Liu Y B, Wang N, Hao Z F, San Vicente F, Olsen M S, Prasanna B M, Lu Y L, Zhang X C. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize., 2019, 30:1919.
[10] Wang X T, Wu L J, Zhang S F, Wu L C, Ku L X, Wei X M, Xie L L, Chen Y H. Robust expression and association ofwith circadian rhythms in maize., 2011, 30: 1261–1272.
[11] Alter P, Bircheneder S, Zhou L Z, Schlüter U, Gahrtz M, Sonnewald U, Dresselhaus T. Flowering time-regulated genes in maize include the transcription factor., 2016, 172: 389–404.
[12] Jin M L, Liu X G, Jia W, Liu H J, Li W Q, Peng Y, Du Y F, Wang Y B, Yin Y J, Zhang X H, Liu Q, Deng M, Li N, Cui X Y, Hao D Y, Yan J B., agene represses flowering in maize by interfering with the circadian clock and activating expression of., 2018, 60: 465–480.
[13] Muszynski M G, Dam T, Li B L, Shirbroun D M, Hou Z L, Bruggemann E, Archibald R, Ananiev E V, Danilevskaya O N.encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize., 2006, 142: 1523–1536.
[14] Colasanti J, Tremblay R, Wong A Y, Coneva V, Kozaki A, Mable B K. The maizeflowering time regulator defines a highly conserved zinc finger protein family in higher plants., 2006, 7: 158.
[15] Salvi S, Tuberosa R, Chiapparino E, Maccaferri M, Veillet S, Van Beuningen L, Isaac P, Edwards K, Phillips R L. Toward positional cloning of, a QTL controlling the transition from the vegetative to the reproductive phase in maize., 2002, 48: 601–613.
[16] Guo L, Wang X H, Zhao M, Huang C, Li C, Li D, Yang C J, York A M, Xue W, Xu G H, Liang Y, Chen Q Y, Doebley J F, Tian F. Stepwise cis-regulatory changes incontribute to maize flowering-time adaptation., 2018, 28: 3005–3015.
[17] Liang Y M, Liu Q, Wang X F, Huang C, Xu G H, Hey S, Lin H Y, Li C, Xu D Y, Wu L S, Wang C L, Wu W H, Xia J L, Han X, Lu S J, Lai J S, Song W B, Schnable P S, Tian F.functions as a flowering activator through the.-regulatory module and contributes to maize flowering time adaptation., 2019, 221: 2335–2347.
[18] Huang C, Sun H Y, Xu D Y, Chen Q Y, Liang Y M, Wang X F, Xu G H, Tian J G, Wang C L, Li D, Wu L S, Yang X H, Jin W W, Doebley J F, Tian F.enhances maize adaptation to higher latitudes., 2018, 115: E334–E341.
[19] Hung H Y, Shannon L M, Tian F, Bradbury P J, Chen C, Flint-Garcia S A, Mcmullen M D, Ware D, Buckler E S, Doebley J F, Holland J B.and the genetic basis of day-length adaptation underlying the postdomestication spread of maize., 2012, 109: E1913–E1921.
[20] 姜洪真, 马伯军, 钱前, 高振宇. 全基因组关联分析(GWAS)在作物农艺性状研究中的应用. 农业生物技术学报, 2018, 26: 1244–1257. Jiang H Z, Ma B J, Qian Q, Gao Z Y. The application of genome-wide association study (GWAS) in crop agronomic traits., 2018, 26: 1244–1257 (in Chinese with English abstract).
[21] 杨宇昕, 桑志勤, 许诚, 代文双, 邹枨. 利用WGCNA进行玉米花期基因共表达模块鉴定. 作物学报, 2019, 45: 161–174. Yang Y X, Sang Z Q, Xu C, Dai W S, Zou C. Identification of maize flowering gene co-expression modules by WGCNA., 2019, 45: 161–174 (in Chinese with English abstract).
[22] 邓照, 蒋环琪, 程丽沙, 刘睿, 黄敏, 李曼菲, 杜何为. 利用WGCNA鉴定玉米非生物胁迫相关基因共表达网络. 作物学报, 2023, 49: 672–685. Deng Z, Jiang H Q, Cheng L S, Liu R, Huang M, Li M F. Identification of abiotic stress-related gene co-expression networks in maize by WGCNA., 2023, 49: 672–685 (in Chinese with English abstract).
[23] Stelpflug S C, Sekhon R S, Vaillancourt B, Hirsch C N, Buell C R, De Leon N, Kaeppler S M. An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development., 2016, 9, 1–16.
[24] Knapp S J, Stroup W W, Ross W M. Exact confidence intervals for heritability on a progeny mean basis., 1985, 25: 192–194.
[25] 刘小磊. 一种交替运用固定效应和随机效应模型优化全基因组关联分析的算法开发. 华中农业大学博士学位论文, 湖北武汉, 2016. Liu X L. Development of an Iterative Usage of Fixed Effect and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PhD Dissertation of Huazhong Agricultural University, Wuhan, Hubei, China, 2016 (in Chinese with English abstract).
[26] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[27] 鲜小华, 王嘉, 徐新福, 曲存民, 卢坤, 李加纳, 刘列钊. 整合GWAS和WGCNA分析挖掘甘蓝型油菜黄籽微效作用位点. 作物学报, 2018, 44: 1105–1113. Xian X H, Wang J, Xu X F, Qu C M, Lu K, Li J N, Liu L D. Mining yellow-seeded micro effect loci inby integrated GWAS and WGCNA analysis., 2018, 44: 1105–1113 (in Chinese with English abstract).
[28] Downs G S, Bi Y M, Colasanti J, Wu W Q, Chen X, Zhu T, Rothstein S J, Lukens L N. A developmental transcriptional network for maize defines co-expression modules., 2013, 161: 1830–1843.
[29] Sheehan M J, Kennedy L M, Costich D E, Brutnell T P. Subfunctionalization ofandin the control of seedling and mature plant traits in maize., 2007, 49: 338–353.
[30] Barnes A C, Rodríguez-Zapata F, Juárez-Núñez K A, Gates D J, Janzen G M, Kur A, Wang L, Jensen S E, Estévez-Palmas J M, Crow T M, Kavi H S, Pil H D, Stokes R L, Knizner K T, Aguilar-Rangel M R, Demesa-Arévalo E, Skopelitis T, Pérez-Limón S, Stutts W L, Thompson P, Chiu Y C, Jackson D, Muddiman D C, Fiehn O, Runcie D, Buckler E S, Ross-Ibarra J, Hufford M B, Sawers R J H, Rellán-Álvarez R. An adaptive teosinte mexicana introgression modulates phosphatidylcholine levels and is associated with maize flowering time., 2022, 119: e2100036119.
[31] Bendix C, Mendoza J M, Stanley D N, Meeley R, Harmon F G. The circadian clock-associated geneaffects maize developmental transitions., 2013, 36: 1379–1390.
[32] Hayes K R, Beatty M, Meng X, Simmons C R, Habben J E, Danilevskaya O N. Maize global transcriptomics reveals pervasive leaf diurnal rhythms but rhythms in developing ears are largely limited to the core oscillator., 2010, 5: e12887.
[33] Castelletti S, Tuberosa R, Pindo M, Salvi S. A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL., 2014, 4: 805–812.
[34] Liu L, Wu Y, Liao Z, Xiong J, Wu F, Xu J, Lan H, Tang Q, Zhou S, Liu Y, Lu Y. Evolutionary conservation and functional divergence of the LFK gene family play important roles in the photoperiodic flowering pathway of land plants., 2018, 120: 310–328.
[35] Li Q L, Liu B S. Genetic regulation of maize flower development and sex determination., 2017, 245:1–14.
[36] Li D, Wang X F, Zhang X B, Chen Q Y, Xu G H, Xu D Y, Wang C L, Liang Y M, Wu L S, Huang C, Tian J G, Wu Y Y, Tian F. The genetic architecture of leaf number and its genetic relationship to flowering time in maize., 2016, 210: 256–268
[37] Mascheretti I, Battaglia R, Mainieri D, Altana A, Lauria M, Rossi V. The WD40-repeat proteinsandregulate different aspects of maize development through chromatin modification., 2013, 25: 404–420.
[38] Pandey P, Srivastava P K, Pandey S P. Prediction of plant miRNA targets., 2019, 1932: 99–107.
[39] Ligaba-Osena A, Dimarco K, Richard T L, Hankoua B. The maizemiRNA-regulated developmental alterations are restored by a bacterial ADP-glucose pyrophosphorylase in transgenic tobacco., 2018, 2018: 8581258.
[40] Piñeiro M, Gómez-Mena C, Schaffer R, Martínez-Zapater J M, Coupland G. Early bolting in short days is related to chromatin remodeling factors and regulates flowering inby repressing., 2003, 15: 1552–1562
[41] Mccormick A J, Kruger N J. Lack of fructose 2,6-bisphosphate compromises photosynthesis and growth inin fluctuating environments., 2015, 81: 670–683.
[42] 钱景华, 李增强, 廖小芳, 汤丹峰, 史奇奇, 周瑞阳, 陈鹏. 调控植物花发育的MYB类转录因子研究进展. 生物技术通讯, 2016, 27: 283–288.Qian J H, Li Z Q, Liao X F, Tang D F, Shi Q Q, Zhou R Y, Chen P. Advance on MYB transcription factors in regulating plant flower development., 2016, 27: 283–288 (in Chinese with English abstract).
[43] Zhang X B, Chen Y H, Wang Z Y, Chen Z L, Gu H Y, Qu L J. Constitutive expression of() affects several circadian-regulated processes and seed germination in., 2007, 51: 512–525.
[44] Barth C, Tullio M D, Conklin P L. The role of ascorbic acid in the control of flowering time and the onset of senescence., 2006, 57: 1657–1665.
[45] Liu H Y, Zhou X C, Li Q P, Wang L, Xing Y Z. CCT domain-containing genes in cereal crops: flowering time and beyond., 2020, 133: 1385–1396.
[46] Cardona-López X, Cuyas L, Marín E, Rajulu C, Irigoyen M L, Gil E, Puga M I, Bligny R, Nussaume L, Geldner N, Paz-Ares J, Rubio V. ESCRT-III-associated proteinmediates high- affinity phosphate transporter trafficking to maintain phosphate homeostasis in., 2015, 27: 2560–2581.
[47] Mai Y X, Wang L, Yang H Q. A gain-of-function mutation inconfers late flowering under short-day light in., 2011, 53: 480–492.
[48] Doukhanina E V, Chen S R, Van Der Zalm E, Godzik A, Reed J, Dickman M B. Identification and functional characterization of the BAG protein family in., 2006, 281: 18793–18801.
[49] Huang J, Sun W, Ren J X, Yang R C, Fan J S, Li Y F, Wang X, Joseph S, Deng W B, Zhai L H. Genome-wide identification and characterization of actin-depolymerizing factor (ADF) family genes and expression analysis of responses to various stresses inL., 2020, 21: 1751.
[50] Yu Y C, Qiao L F, Chen J C, Rong Y H, Zhao Y H, Cui X K, Xu J P, Hou X M, Dong C H.acts as a B3 domain transcription factor to promote flowering time via directly binding to the promoters ofand., 2020, 103: 1386–1398.
[51] 邢瑞霞, 朱金洁, 祁显涛, 谢传晓, 江海洋, 刘昌林. 玉米开花期调控机制研究进展. 安徽农业科学, 2022, 50(9): 23–26. Xing R X, Zhu J J, Qi X T, Xie C X, Jiang H Y, Liu C L. Research progress on the regulation mechanism of maize flowering period., 2022, 50(9): 23–26 (in Chinese with English abstract).
[52] Xu J, Liu Y X, Liu J, Cao M J, Wang J, Lan H, Xu Y B, Lu Y L, Pan G T, Rong T Z. The genetic architecture of flowering time and photoperiod sensitivity in maize as revealed by QTL review and meta analysis., 2012, 54: 358–373.
[53] Wang L W, Zhou Z Q, Li R G, Weng J F, Zhang Q G, Li X H, Wang B Q, Zhang W Y, Song W, Li X H. Mapping QTL for flowering time-related traits under three plant densities in maize., 2021, 9: 372–379.
[54] Khairallah M M, Bohn M, Jiang C, Deutsch J A, Jewell D C, Mihm J A, Melchinger A E, González-De-León D, Hoisington D A. Molecular mapping of QTL for southwestern corn borer resistance, plant height and flowering in tropical maize., 1998, 117: 309–318.
[55] 侯清桂, 张君, 田磊, 徐梦真, 邹欢, 毛棣, 陈彦惠, 吴连成. 基于SNP标记连锁图谱的玉米花期性状QTL定位. 玉米科学, 2021, 29(6): 41–49. Hou Q G, Zhang J, Tian L, Xu M Z, Zou H, Mao L, Chen Y H, Wu L C. QTL mapping of maize flowering traits based on SNP molecular maker linkage map., 2021, 29(6): 41–49 (in Chinese with English abstract).
[56] 杨慧丽, 林亚楠, 张怀胜, 卫晓轶, 丁冬, 薛亚东. 玉米开花期性状的QTL及杂种优势位点定位. 作物学报, 2017, 43: 678–690. Yang H L, Lin Y N, Zhang H S, Wei X T, Ding D, Xue Y D. Mapping of QTLs and heterotic loci for flowering time-related traits in maize., 2017, 43: 678–690 (in Chinese with English abstract).
[57] 袁亮, 孟鑫, 汪亚龙, 廖长见, 李高科, 吕桂华, 宋军, 邱正高,林海建. 镉胁迫下甜、糯玉米开花期性状的全基因组关联分析.植物遗传资源学报, 2021, 22: 438–447. Yuan L, Meng X, Wang Y L, Liao C J, Li G K, Lyu G H, Song J, Qiu Z G, Lin H J. Genome wide association analysis of flowering traits in sweet and waxy maize under cadmium stress., 2021, 22: 438–447 (in Chinese with English abstract).
[58] Shi J, Wang Y H, Wang C H, Wang L, Zeng W, Han G M, Qiu C H, Wang T Y, Tao Z, Wang K J, Huang S J, Yu S S, Wang W Y, Chen H Y, Chen C, He C, Wang H, Zhu P L, Hu Y Y, Zhang X, Xie C X, Lu X D, Li P J. Linkage mapping combined with GWAS revealed the genetic structural relationship and candidate genes of maize flowering time-related traits., 2022, 22: 328.
[59] 魏海忠, 商伟, 钟世宜, 张彦军, 徐长利, 赵燕, 王红红, 刘保申. 利用重组自交系群体定位玉米生育期相关性状QTL. 玉米科学, 2014, 22(1): 49–55. Wei H Z, Shang W, Zhong S W, Zhang Y J, Zhao Y, Wang H H, Liu B S. Mapping of growth period related traits in maize using recombinant inbred lines., 2014, 22(1): 49–55 (in Chinese with English abstract)).
[60] 李凯, 姜涛, 才源, 王丕武, 陈雪峰, 马科, 周元元, 卢石. 玉米花期性状的主效SSR标记筛选. 玉米科学, 2015, 23(1): 33–38.Li K, Jiang T, Cai Y, Wang P W, Chen X F, Ma K, Zhou Y Y, Lu S. Screening of the main effect SSR markers of maize flowering., 2015, 23(1): 33–38 (in Chinese with English abstract).
[61] 郭向阳, 陈建军, 卫晓轶, 祝云芳, 王安贵, 刘鹏飞, 汤继华, 陈泽辉. 施氮与不施氮条件下玉米开花期相关性状的QTL定位. 植物营养与肥料学报, 2017, 23: 297–303. Guo X Y, Chen J J, Wei X Y, Zhu Y F, Wang A G, Liu P F, Tang J H, Chen Z H. QTL mapping of flowering related traits of maize with and without nitrogen application., 2017, 23: 297–303 (in Chinese with English abstract).
[62] 何文昭, 王红武, 胡小娇, 李坤, 王琪, 吴宇锦, 刘志芳, 黄长玲. 玉米株高和穗位高在不同环境下的数量遗传分析. 作物杂志, 2017, (3): 13–18.He W Z, Wang H W, Hu X J, Li K, Wang Q, Wu Y J, Liu Z F, Huang C L. Quantitative genetic research of plant height and ear height in maize under different environments., 2017, (3): 13–18 (in Chinese with English abstract).
[63] 曾群, 赵仲华, 赵淑清. 植物开花时间调控的信号途径. 遗传, 2006, 28: 1031–1036.Zeng Q, Zhao Z H, Zhao S Q. Signal pathways of flowering time regulation in plant., 2006, 28: 1031–1036 (in Chinese with English abstract).
[64] Huang D M, Lin W F, Deng B, Ren Y J, Miao Y. Dual-locatedinteracting withalters photochemical activities of photosystem I and is involved in light adaptation in., 2017, 18: 2352.
[65] Su H H, Liang J C, Abou-Elwafa S F, Cheng H Y, Dou D D, Ren Z Z, Xie J R, Chen Z H, Gao F G, Ku L X, Chen Y H.regulates photoperiod-dependent flowering and response to stresses in maize., 2021, 21: 453.
[66] Guo J, Li C H, Zhang X Q, Li Y X, Zhang D F, Shi Y S, Song Y C, Li Y, Yang D G, Wang T Y. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress., 2020, 292: 110380.
[67] Ma L L, Zhang M Y, Chen J, Qing C Y, He S J, Zou C Y, Yuan G S, Yang C, Peng H, Pan G T, Lübberstedt T, Shen Y. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (L.) seedlings., 2021, 134: 3305–3318.
[68] 王艳花, 刘景森, 李加纳. 整合GWAS和WGCNA筛选鉴定甘蓝型油菜生物产量候选基因. 作物学报, 2021, 47: 1491–1510. Wang Y H, Liu J S, Li J N. Integrating GWAS and WGCNA to screen and identify candidate genes for biological yield inL., 2021, 47: 1491–1510 (in Chinese with English abstract).
[69] Francisco F R, Aono A H, Da Silva C C, Gonçalves P S, Scaloppi Junior E J, Le Guen V, Fritsche-Neto R, Souza L M, De Souza A P. Unravelling rubber tree growth by integrating GWAS and biological network-based approaches., 2021, 12: 768589.
[70] Raman H, Raman R, Coombes N, Song J, Prangnell R, Bandaranayake C, Tahira R, Sundaramoorthi V, Killian A, Meng J, Dennis E S, Balasubramanian S. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola., 2016, 39: 1228–1239.
[71] Hartmann U, Höhmann S, Nettesheim K, Wisman E, Saedler H, Huijser P. Molecular cloning of: a negative regulator of the floral transition in., 2000, 21: 351–360.
[72] 王佳丽, 王鹤冰, 杨慧勤, 胡若琳, 魏大勇, 汤青林, 王志敏. NAC转录因子在植物花发育中的作用. 生物工程学报, 2022, 38: 2687–2699. Wang J L, Wang H B, Yang H Q, Hu R L, Wei D Y, Tang Q L, Wang Z M. The role of NAC transcription factors in flower development in plants., 2022, 38: 2687–2699 (in Chinese with English abstract).
[73] Kim S G, Kim S Y, Park C M. A membrane-associated NAC transcription factor regulates salt-responsive flowering viain., 2007, 226: 647–654.
[74] 陈旭. NAC家族转录因子介导赤霉素信号通路参与调控水稻株高和开花时间. 复旦大学博士学位论文, 上海, 2013. Chen X. Expression of Rice NAC Transcription FactorReduced the Height of Rice and Delayed the Flowering Time by Gibberellin Pathway. PhD Dissertation of Graduate School of Fudan University, Shanghai, China, 2013 (in Chinese with English abstract).
[75] Pimenta M R, Silva P A, Mendes G C, Alves J R, Caetano H D, Machado J P, Brustolini O J, Carpinetti P A, Melo B P, Silva J C, Rosado G L, Ferreira M F, Dal-Bianco M, Picoli E A, Aragao F J, Ramos H J, Fontes E P. The stress-induced soybean NAC transcription factorplays a positive role in developmentally programmed leaf senescence., 2016, 57: 1098–1114.
[76] Du Y F, Lunde C, Li Y F, Jackson D, Hake S, Zhang Z X. Gene duplication at thelocus controls the fate of inflorescence meristem cells in maize., 2021, 118: e2019218118.
Mining maize flowering traits related candidate genes based on GWAS and WGCNA data
QIAN Fu1, ZHANG Zhan-Qin2, CHEN Shu-Bin2, DING Yong-Fu2, SANG Zhi-Qin2,*, and LI Wei-Hua1,*
1College of Agriculture, Shihezi University / Key Laboratory of Oasis Eco-Agriculture, Shihezi 832000, Xinjiang, China;2Xinjiang Academy of Agricultural and Reclamation Science, Shihezi 832000, Xinjiang, China
The flowering time is one of the important traits in maize. It is of great significance to analyze the genetic basis and to mine the key core genes in flowering for maize varieties with wide adaptability. A natural population consisting of 580 maize inbred lines were planted for three years, to determine the three flowering traits (including days to anthesis, days to silking, and anthesis silking interval). Genome-wide association analysis was conducted using 31,826 SNPs distributed throughout the whole genome. Combined with transcriptome data of 14 different developmental stages of inbred line B73, weighted gene co-expression network analysis was performed to select tissue specific modules and key genes related to maize flowering time. A total of 14 SNPs for four flower traits under multiple environments and 10 potential candidate genes were mined by GWAS, WGCNA was used to mine 17 potential candidate genes in flowering time, three candidate genes were jointly mined by both approaches.encodes a MADS box transcription factor 19,encodes the NAC transcription factor 133,encodes MADS box transcription factor 8, mainly involved in regulating inflorescence growth and development, which has certain research value and significance. These results provide a reference for the genetic basis and molecular mechanisms of flowering time related traits in maize.
maize; flowering time related traits; GWAS, WGCNA; candidate gene
2023-05-24;
2023-06-09.
10.3724/SP.J.1006.2023.33010
通信作者(Corresponding author): 李卫华, E-mail: lwh_agr@shzu.edu.cn; 桑志勤, E-mail: sangzhiqin@126.com
E-mail: 1971194140@qq.com
2023-02-22;
本研究由兵团重点领域科技攻关项目(2019AB021), 兵团科技创新人才计划项目(2021CB038)和中国科学院“西部青年学者”项目资助。
This study was supported by the Tackling Key Scientific and Technological Problems in Key Areas of Xinjiang Production and Construction Corps (2019AB021), the Science and Technology Innovation Talent Plan of Xinjiang Production and Construction Corps (2021CB038), and the Chinese Academy of Sciences “Western Young Scholar”.
URL: https://kns.cnki.net/kcms2/detail/11.1809.S.20230608.1057.004.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
中学生天地(A版)(2023年1期)2023-02-17
今日农业(2021年15期)2021-10-14
商品与质量(2021年16期)2021-04-23
意林(2018年18期)2018-09-30
生命科学研究(2018年1期)2018-05-29
作文周刊·小学一年级版(2017年35期)2017-10-18
上海农业学报(2017年3期)2017-04-10
山东农业工程学院学报(2016年6期)2016-12-01
传奇故事(破茧成蝶)(2015年8期)2015-02-28
