
HPLC法测定食品中脱氢乙酸含量的方法优化
2023-11-07 11:45:58范灵慧
食品工业 2023年10期
范灵慧
上海源本食品质量检验有限公司(上海 200090)
脱氢乙酸,固态为白色或略微淡黄色具有结晶性的颗粒状粉末,无臭、没有任何气味、熔点108~110℃,沸点270 ℃,是一种具有低毒、高效的防腐、防霉剂。在酸、碱等环境下具有一定的抗菌功能,特别是对霉菌的抑制作用最强[1]。它通常是一种有效的消毒剂。但是过量服用会对身体产生一定的危害。我国允许在部分食品中添加使用。GB 5009.121—2016《食品安全国家标准 食品中脱氢乙酸的测定》中脱氢乙酸的液相色谱法前处理过程较为复杂,不同基质处理方法不同,进样后色谱峰拖尾严重,此次试验在国标基础上进行简化,优化流动相比例和不同基质的前处理方法,使实验操作更简单且峰型更对称。
1 材料与方法
1.1 仪器与试剂
液相色谱仪Ultimate3000(赛默飞世尔科技有限公司);电子天平BSA 423S(德国赛多利斯公司);超声波清洗器FXP 20M(Unisonics Australia);涡旋震荡混合器BE-2600(其林贝尔仪器制造有限公司);实验室pH计FE20[梅特勒-托利多仪器(上海)有限公司]。
乙酸铵[默克化工技术(上海)有限公司];氢氧化钠(永华化学股份有限公司);硫酸锌(上海凌峰化学试剂有限公司);甲醇(色谱纯,美国默克公司);氨水(永华化学股份有限公司);超纯水(上海和泰仪器有限公司实验室超纯水机制得);0.45 μm水相滤膜(上海安谱实验科技股份有限公司);脱氢乙酸(dehydroacetic acid)标准品(BePure,纯度大于99.9%)。
标准储备液:准确称取0.050 g脱氢乙酸标准品,用20 g/L的氢氧化钠溶液溶解后用水定容至50 mL,配制成质量浓度1.0 mg/mL的标准储备液。
标准工作液:标准储备液经逐级稀释后,配制成1,10.0,50.0,100和200 μg/mL标准工作液质量浓度。检测用食品:市场流通渠道购买。
1.2 仪器工作条件
色谱柱DikMA Silversil 5 μm C18,250 mm×4.6 mm;柱温35 ℃;进样量10 μL;流速1.0 mL/min;检测波长293 nm;进样时间22 min;流动相为乙酸铵溶液(0.02 mol/L)-甲醇,洗脱比例见表1。

表1 脱氢乙酸流动相梯度洗脱比例
1.3 样品前处理
称取2 g试样于50 mL玻璃比色管中,加入25 mL超纯水,旋涡混合1 min,超声提取30 min以上,取出后再次涡旋混合1 min,加入5 mL的ZnSO4溶液(120 g/L),用NaOH溶液(20 g/L)调节pH 7.5,用水定容至50 mL,摇匀,用滤纸过滤,取澄清滤液,过0.45 μm的水相滤膜后上机测定。
2 结果与讨论
2.1 前处理中蛋白沉淀剂的选择
分别对常用的无机沉淀蛋白体系,即亚铁氰化钾和乙酸锌、亚铁氰化钾和硫酸锌、氢氧化钠和硫酸锌进行加标回收试验。结果表明,在3种沉淀体系中氢氧化钠与硫酸锌沉淀法的回收率最高,故试验选用氢氧化钠和硫酸锌作为蛋白沉淀剂。
2.2 色谱操作条件的确定
2.2.1 检测波长的确定
使用DAD检测器,在3D光谱图上读取到脱氢乙酸吸收峰最大时候的波长分别在230 nm和293 nm处,但是在230 nm处时,杂质干扰多,在293 nm处杂质干扰少,故选取检测波长293 nm,与国标方法波长一致。
2.2.2 流动相梯度洗脱比例
脱氢乙酸抗酵母菌和霉菌的效果较好,因此被广泛应用于面包、糕点和发酵食品中,由于这些食品中经常会同时添加各种类型的添加剂,为使样品中性质差异较大的组分能达到良好的分离效果,以及洗脱干净的目的,试验采用梯度洗脱。洗脱比例见表1。
2.2.3 流动相pH的选择
在相同的仪器条件下,对质量浓度50.0 μg/mL的脱氢乙酸标品进样后对比图谱。使用GB 5009.121—2016《食品安全国家标准 食品中脱氢乙酸的测定》中方法,0.02 mol/mL乙酸铵-甲醇9∶1(体积比)流动相进样图谱如图1所示。试验将0.02 mol/mL的乙酸铵用氨水调节pH 9.0,初始流动相比例为甲醇-0.02 mol/mL乙酸铵1∶49(体积比),并采用表1梯度洗脱,进样图谱如图2所示。
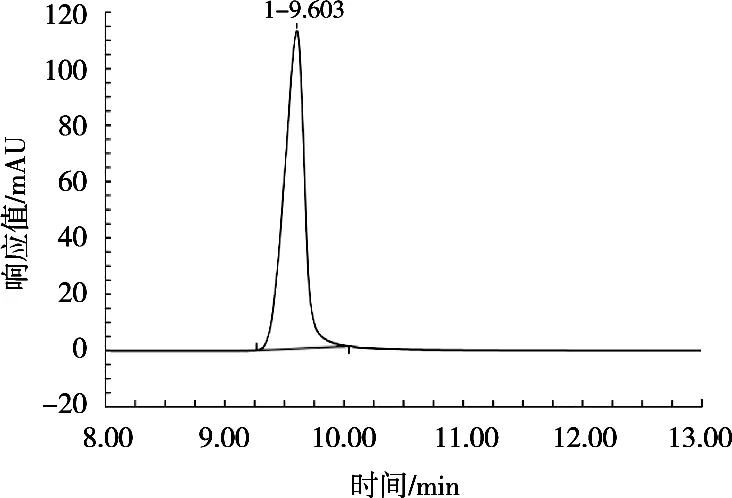
图1 0.02 mol/mL乙酸铵与甲醇9∶1(体积比)流动相进样图谱
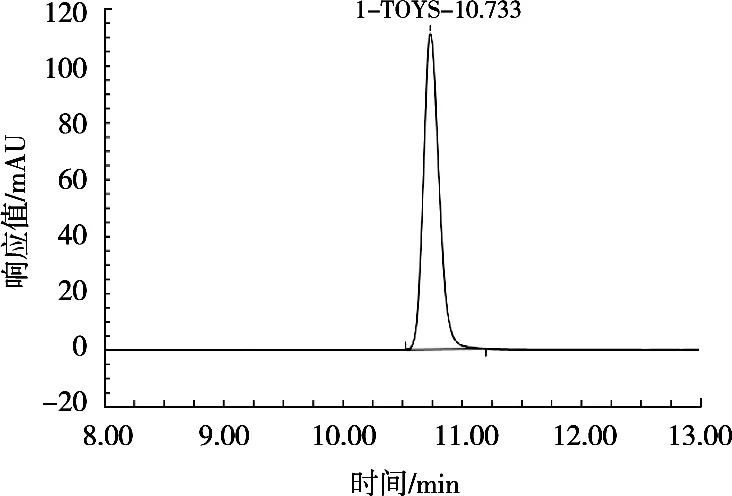
图2 用氨水调节pH的0.02 mol/mL乙酸铵与甲醇1∶49(体积比)进样图谱
由图1和图2的图谱对比可看出,使用试验优化后的流动相进样,得到的脱氢乙酸色谱峰的峰型和对称性较好。
2.3 线性关系
标准品质量浓度在1.0~200.0 mg/mL的质量浓度范围内响应值好,峰面积的线性关系良好,线性方程y=0.482 8x,相关系数R2=0.999 7。见表2。

表2 脱氢乙酸检出限、定量限及线性方程
2.4 回收试验
2.4.1 国标法与试验优化方法回收率比较
根据GB 5009.121—2016 《食品安全国家标准 食品中脱氢乙酸的测定》第二法液相色谱法中12.1试样制备与提取可知,国标法对不同的基质有不同的前处理方式。试验简化后的前处理方法则可统一处理不同基质的样品,选用果蔬汁、腐乳、糕点和黄油作为试验基质加入脱氢乙酸标准品,分别按照国标方法和试验方法前处理后进样,对比它们的加标回收率,结果见表3。

表3 不同基质样品不同前处理方法回收率
2.4.1.1 果蔬汁
按国标方法称样在离心管中,需要调pH后转移到容量瓶中定容,置于离心管中离心取上清液调节pH后再次定容,取5 mL过固相萃取柱,收集洗脱液后上机测定,在该前处理过程中,反复的转移和过固相萃取柱都会造成损失。而试验优化后的前处理直接称样在比色管中,加入沉淀剂后用比色管定容即可,减少反复转移和固相萃取柱净化的步骤,可以提高回收率。由数据结果也可得出国标方法回收率较优化后方法低。
2.4.1.2 腐乳
按国标方法称样在离心管中,转移到容量瓶中定容,超声提取后过滤上机,回收率达99%,试验前处理优化后回收率也达100%,在回收率相差不大的情况下,试验的前处理过程更为简单和方便。
2.4.1.3 糕点
按国标法称样溶解加入沉淀剂定容后需要转移至分液漏斗中,用正己烷重复抽提后取水相层离心,取上清液过滤膜测定,若高效液相色谱分离效果不明显,需要取上清液调节pH后过固相萃取柱,洗脱后再上机。从表3可知,通过国标法萃取后,回收率已较低,过固相萃取柱之后损失更大,而用试验优化后的前处理方法,回收率较好。
2.4.1.4 黄油
根据国标法需要用正己烷抽提后离心上机,若分离效果不佳再过固相萃取柱净化,试验国标法回收率达103%,但是过固相萃取柱之后回收率为98%,而试验回收率为109%,虽然操作便捷,但是试验优化后减少去油脂的步骤,对油脂含量高的食品检测结果影响较大,故回收效果不如国标方法。
因此,试验优化后的前处理方法适用于大部分的基质样品,进样后分离度好,且回收率高。国标方法若是分离效果不明显需要再次过固相萃取柱,造成较大损失。但当样品为油类基质时,更推荐使用国标法。
2.4.2 方法优化后回收率与精密度
选取蔬菜汁、腐乳、糕点样品,按定量限的3,5和10倍范围进行加标回收,故分别在0.015 0,0.025 0和0.050 0 g/kg三水平添加脱氢乙酸,按优化的前处理方法进行提取和测定,每个添加水平平行测定6次,计算回收率和精密度。试验结果表明,测得的平均回收率范围为97.1%~104.0%,SRSD在0.34%~2.32%。测定结果见表4。
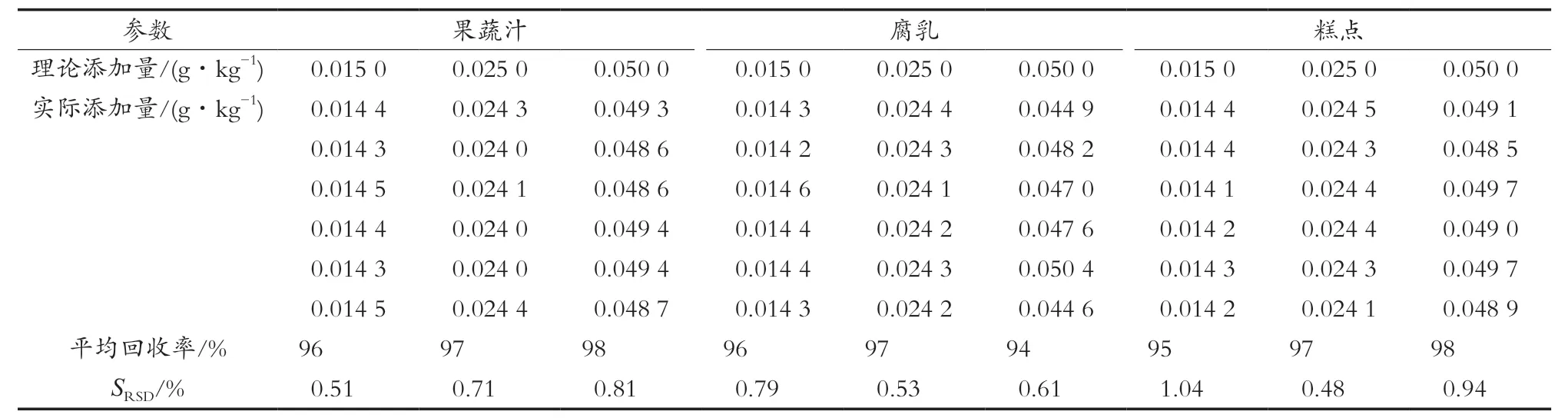
表4 添加回收率和精密度(n=6)

表5 糕点中的脱氢乙酸质控回收率数据
2.4.3 实物质控样
质控样品为糕点中的脱氢乙酸,经过试验的前处理步骤后,上机测定值为0.259 g/kg,折算质控参考值0.265 g/kg,回收率达97.8%。按照试验方法检测结果为满意,数据结果证实方法的准确性好。
3 结论
试验针对食品中脱氢乙酸的提取和分析条件在国标的基础上进行优化,利用高效液相色谱仪对其进行有效的分离和测定,该方法具有较好的精密度和回收率,线性关系良好,针对不同基质也可使用同样的前处理方法,实现不同基质样品可同时处理,对检测任务较重的第三方检测可以节约大量时间成本和人力成本。
猜你喜欢
食品安全导刊(2020年21期)2020-09-07 09:14:04
中国油脂(2020年3期)2020-04-10 02:08:54
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
中国自行车(2018年3期)2018-04-18 07:16:33
无机化学学报(2016年8期)2016-12-06 09:05:14
汽车与安全(2016年5期)2016-12-01 05:22:15
山东工业技术(2016年13期)2016-06-29 09:05:13
陕西教育·综合版(2016年11期)2016-04-12 09:10:31
化学分析计量(2016年1期)2016-03-14 00:35:19
