
星形胶质细胞在阿尔茨海默病中的研究进展
2023-11-01 06:21:52胡承平陶凤芝秦虹云
同济大学学报(医学版) 2023年5期
胡承平, 陶凤芝, 王 灿, 秦虹云
(同济大学附属精神卫生中心精神科,上海 200124)
阿尔茨海默病(Alzheimer’s disease, AD)是一种慢性进行性神经系统变性疾病,以进行性、不可逆性记忆障碍及其他高级认知功能损害为主要临床表现[1]。其病因和发病机制非常复杂,主要包括细胞外β淀粉样蛋白(amyloid-β, Aβ)沉积、细胞内tau蛋白异常聚集、胆碱能系统失调、突触受损、中间神经元异常、脑网络异常、基因突变、免疫学异常等[2-4]。其中,淀粉样级联假说认为,Aβ沉积触发一系列级联反应,导致神经元变性,是AD出现认知功能障碍的主要原因[5]。中枢神经系统(central nervous system, CNS)中维持内稳态的胶质细胞如星形胶质细胞、少突胶质细胞、NG2胶质细胞、小胶质细胞等均参与这个过程[6],尤其是,体积最大,数量最多的星形胶质细胞(astrocyte, AS)[5]。
AS于1893年首次被Michael von Lenhossek命名[7],生理情况下,具有营养支持功能,分泌生长因子(如EGF、FGF、IGF、CNTF)等营养神经元[8-9],通过糖酵解途径产生乳酸和转运葡萄糖为神经元提供能量支持[5,10];具有物理屏障和调节脑血流的功能,参与血脑屏障[6,11]和神经血管单元(neurovascular unit, NVU)[12]的形成;具有免疫调节功能,分泌炎症因子产生神经炎症反应[13-15];具有维持神经递质、细胞外离子和水等的稳态功能[6];具有吞噬功能,参与突触的形成和修剪[13-15]。
在病理因素作用下,AS主要表现为3种变化,即萎缩、变性和增殖[16]。其中增殖也被称为反应性星形胶质细胞(reactive astrocytes, RAS)。RAS大致可分为2种类型,分别为分泌神经营养因子促进神经修复的A2s和对神经元产生毒害作用的A1s。其中,与AD等神经退行性疾病密切相关的是萎缩的AS[6,16]和A1s[17-19]。因此,AS通过正常生理功能的减弱,病理功能的增强来影响脑内神经元的结构和功能,其在AD发生发展过程中发挥着重要作用[7,20-22]。本文将综合已有的研究,探讨AS在AD发生发展过程中的作用。
1 AS与脑内能量代谢
AS是神经元能量代谢的供给者。AS用终足包裹微血管,组成神经血管单元(neurovascular unit, NVU)[12],与神经元广泛联系组成星形胶质细胞-神经元乳酸穿梭单元(astrocyte-neuron lactate shuttle, ANLS),以此来调节血流量和神经细胞的能量供应[8]。AS从血液中摄取葡萄糖,进行糖酵解产生乳酸,通过单羧酸转运体和葡萄糖转运体GLUT1[23]将葡萄糖供给神经元来获取能量(astrocyte-neuron lactate shuttle, ANLS)[5,10],来保障没有糖原储备的神经元对能量的大量需求。病理检查发现AD患者脑组织中的葡萄糖浓度升高,而糖酵解相关的酶,葡萄糖转运体GLUT1和GLUT3均较正常对照者下降[24],与疾病的严重程度也一致,而且主要来源于神经元的GLUT3下降更明显,这些均与[18F]FDG-PET检查表现的脑代谢能力,也就是摄取葡萄糖能力下降相一致[5]。在APP/PS1转基因小鼠[25]中发现,GLUT1的下降与Aβ的大量沉积,海马萎缩相一致。因此,在AD病变过程中,神经元本身葡萄糖转运能力下降的同时,AS对葡萄糖摄取和糖酵解能力的下降,又进一步加剧了神经元能量供应障碍,形成恶性循环。
2 AS与离子和水平衡
除了能量供给,AS还调节着离子和水的平衡[5-6,26]。在AD病变中,研究比较成熟的AS相关的离子有钾和钙[27]。研究发现,AS细胞膜存在几乎所有的钾离子通道类型,来维持膜转运蛋白所需要的负电位[16,26]。钙离子失衡主要表现为,受体发生变化后,胞内钙离子超载,触发炎症反应和毒性神经递质的释放。AS表面存在瞬时受体电位(transient receptor potential ankyrin 1 channel, TRPA1)是非选择性的钙通道。AD患者脑内TRPA1活性增加,且APP/PS1小鼠脑内TRPA1表达倍增[27]。Lee等[28]的研究表明,Aβ作用于TRPA1受体,导致Ca2+大量内流,激活蛋白磷酸酶2B(protein phosphatase 2B, PP2B),释放大量的促炎因子IL-1β等。Abramov等[29]和Pham等[30]在AS与神经元的共培养体系中发现,Aβ作用于质膜钙ATP酶(plasma membrane Ca2+ATPase, PMCA)和半通道连接蛋白(connexin, CX),同样导致Ca2+超载,不仅消耗了谷胱甘肽,增加了AS和神经元对氧化应激的易感性而导致死亡[29];而且增加了依赖钙与非依赖钙通道的谷氨酸递质释放[30],而钙离子拮抗剂能有效阻断Aβ-Ca2+-谷氨酸释放通路[30],阻止神经元的死亡。
AS通过水通道蛋白(主要为AQP4型)来调节水的平衡[6,27]。AQP4集中在AS的突触周围以及包裹着血管的终足处[27]。病理检查发现,AD患者脑组织血管周围的AQP4表达缺失[31-32]。动物研究则发现[32],敲除Snta1基因的小鼠,示踪剂在脑内CSF流入和间质流出均减慢,同时伴有Aβ的沉积。这就提示,AS在清除Aβ至脑脊液的过程中,依赖AQP4受体调节水平衡来完成。
3 AS与突触减少和神经元信号通路的破坏
AS分泌的胰岛素样生长因子1(insulin-like growth factor 1, IGF1)能够保护神经元免受氧化应激和Aβ沉积的毒害作用[33]。随着AD病变进展,IGF1的保护作用消失,而且AS的功能发生改变,间接损害神经元。AS与神经元的共培养实验证实,Aβ干预24 h后,AS不仅不能清除Aβ,反而是通过大量的钙离子内流、线粒体去极化、氧化应激反应,以及释放活性氧等途径损伤神经元[6,29]。同时,有研究发现,Aβ激活AS后导致其线粒体外膜中的单胺氧化酶B上调,降解了单胺类递质,从而减弱了此类神经递质在突触间的信号传导[34]。
AS包裹着突触,形成三方突触,Aβ对突触的毒性可以通过AS钙超载干扰神经递质的信号传导[35],具体信号通路: Aβ首先与AS细胞膜上P2Y1R或烟碱型胆碱能受体α7受体结合,引起钙离子超载,释放大量有神经毒性作用的胶质递质(钙依赖的谷氨酸递质和/ATP等)到突触间隙中;这些胶质递质作用于突触前膜和后膜神经元上的P2X7Rs或NMDARs受体,增加了神经元的兴奋性和钙超载,干扰了神经递质的释放及信号传导,长此以往导致突触减少和神经元损害。通过药物抑制APP/PS1小鼠脑内[36]AS的钙通道后就能避免神经元受损,且提高其空间记忆成绩。这就提示Aβ沉积,通过激活AS来损害突触结构和神经元[37],可能是AD病变不断进展的一个关键因素。
兴奋性神经递质谷氨酸是脑内最强大的神经毒素,细胞外过量的谷氨酸会引发神经元死亡[6]。生理情况下,谷氨酸由AS通过囊泡释放的方式来分泌[35],通过细胞膜的受体蛋白来重吸收。研究发现,AD患者脑内突出表现为AS的谷氨酸转运蛋白,即兴奋性氨基酸转运体1(glutamate/aspartate transporter, GLAST)和兴奋性氨基酸转运体2(glutamate transporter-1, GLT-1)的功能障碍,提示谷氨酸转运至AS细胞内能力下降[5]。随后在细胞和动物模型中也证实AS的GLAST和GLT-1的表达减少,伴随着脑脊液中谷氨酸水平升高,这就提示,AS的谷氨酸转运异常,导致谷氨酸细胞外聚集,对神经元产生毒性,可能是AD病变的另一个关键因素。
这就说明,与Aβ代谢类似,在AD初期,AS能够有效清除胞外过多的谷氨酸递质,维持突触和神经元正常功能。随着病变进展,AS调节功能失衡,增加了细胞外谷氨酸的释放,产生神经毒性,导致了神经元的死亡。研究还发现,AS可以释放许多经典的补体成分,产生免疫级联反应,导致突触变性,产生神经毒性作用,最终促使神经元的死亡[38]。
4 AS与脑内神经炎症
炎症在AD中的作用愈来愈受到重视[6,39]。临床研究显示,轻度认知功能障碍(mild cognitive impairment, MCI)患者血清或脑脊液中的炎症因子IL-1β增高提示[40-44],在AD临床症状出现前,患者就出现了体内和脑内的炎症反应。具体信号通路为,Aβ激活AS的核因子κB(NF-κB)促进细胞因子基因转录[6],分泌大量促炎细胞因子(如白细胞介素-1β,白细胞介素-6,诱导型一氧化氮合酶、肿瘤坏死因子α),与少突胶质细胞(oligodendrocyte, OL)和小胶质细胞(microglia, MG)等相互作用,加速AD的病变过程[8,45-48]。体外研究证实,MG是通过细胞因子白细胞介素1α(interleukin-1α, IL-1α)、肿瘤坏死因子α(TNF-α)和补体成分亚单位1q这3种成分来诱导AS为A1型AS(A1s)[38]。此外,体内与体外试验均发现,伴随着Aβ沉积和AD进展,MG和AS的TNF-α表达明显增加[49]。以上研究都说明,Aβ激活AS产生大量的炎症反应。
炎症反应是如何导致神经元死亡的呢?一项APP/PSI小鼠的研究发现[50-51],小鼠脑神经元和胶质细胞中存在炎性小体NOD样受体家族1(the NOD-like receptor 1, NLRP1)或NLRP3的高表达,可以导致焦亡。由于焦亡是一种伴随着炎症因子白细胞介素-1β(interleukin-1β, IL-1β)和白细胞介素-18(interleukin-18, IL-18)大量释放的死亡形式,除了炎性小体NLRP1和NLRP3,AS可以通过产生过多的ATP,与MG中的P2X7结合,释放IL-1β。IL-1β等聚集在Aβ斑块附近[52-53],加速Aβ沉积,促进老年斑(senile plaques, SPs)沉积[54]。以上研究说明,Aβ沉积通过诱导AS焦亡或者分泌过多的ATP等途径,导致IL-1β等炎症因子的升高,促进神经元的死亡。
AS除了多途径释放炎症因子以外,还参与各种炎症信号的调节,如上调gp130、TGFβR、IFNγR、ERα、A20、STAT3、FasL和BDNF等信号通路产生抗炎反应,激活Act1、S1P1、B4GALT6、TrκB、NF-κB、SOCS3、CCL2、CXCL10或VEGF等的信号通路发挥促炎作用[55]。在急性病变初期,病变中心促炎信号通路激活为主,而病变周围则表现为抗炎信号通路被激活,增殖的AS使病变局限在有限范围内,随着病程不断进展,脑内微环境发生改变,促炎信号通路进一步被激活,病变范围弥散,表现出了炎症的“双刃剑”特征。这些促炎因子使AS核内诱导型一氧化氮合酶(inducible nitric oxide synthase, iNOS)的表达,释放高浓度的毒性作用的一氧化氮(nitric oxide, NO),促进神经元凋亡和/或坏死[45,56]。然而,AD脑内微环境如何增强AS的促炎信号需要进一步的研究。
一般认为,AD早期病变主要是Aβ沉积,激活CDK-5和GSK-3β,促进tau蛋白磷酸化增多;促进tau蛋白形成寡聚体,激活AS和MG,诱导神经炎症;促进NFTs形成[57]等一系列病理变化,促进AD病程的不断进展,逐渐表现出突触丢失和神经元变性/死亡[58]。在此过程中,AS也会分泌更多的Aβ和P-Tau,进一步加重AD的病理损害。
5 AS与Aβ病理性沉积
在生理情况下AS发挥清除和降解Aβ,阻止Aβ沉积的作用。Wyss-Coray等[59]证实,AS能够降解体外和原位沉积的Aβ。然而,受致病因素的影响,AS这种清除降解能力下降。Simonovitch等[60]就发现,载脂蛋白E4(ApoE4)削弱了AS的自噬作用而导致清除Aβ的能力下降(与ApoE3相比)。此外,有研究还发现,AS的抑制性的调节因子PFKFB3(6-phosphofructo-2-kinase/fructose-2,6-biphosphatase)表达下降,或者增龄等因素[61],导致其糖酵解能力下降以后,能量供应障碍,加剧了Aβ在细胞内和周围积聚[7]。
除了Aβ清除降解减少以外,AS还具有生成Aβ的功能。病理发现,在Aβ斑块周围区域存在着胶质纤维酸性蛋白(glial fibrillary acidic protein, GFAP)增多,说明AS增殖与Aβ沉积密切有关[6]。临床样本中,AD高危人群、MCI(AD前期病变)和AD患者的血浆中,GFAP与脑中Aβ-PET沉积也呈现正相关[62-63],进一步提示,AS增殖与Aβ沉积存在量效关系。转基因小鼠APP/PS1在6月龄脑内出现Aβ沉积[64-65],7-9月龄可观察到明显的AS改变[58]。细胞研究也证实,AS上调了限速酶β分泌酶(β-secretase, BACE1),导致Aβ生成增多[61,66-67]。
以上研究均表明,在AD病程进展中,AS的功能表现为清除降解Aβ的生理功能减弱,病理功能增强如大量生成Aβ,促进其沉积。
6 AS与脑内Tau蛋白的磷酸化
正常老龄人群大脑中就存在AS促进Aβ沉积和P-Tau增加的现象[58]。Aβ沉积通过炎症反应、蛋白酶体失调、自噬损伤、激酶活性增加、磷酸酶活性降低以及轴突运输受阻等方式促进P-Tau的增多[68]。在AD病变过程中,AS导致P-Tau增多更加明显[34]。
MCI和AD患者的血浆GFAP(AS增殖的特征性蛋白)与脑P-Tau表达(PET检查)呈正相关[62]。小鼠模型MAPTP301S也发现,AS增殖的同时P-Tau也明显表达增多[58]。细胞实验进一步证实,P-Tau能诱导AS增生[69],且AS会产生更多的P-Tau(the 3R isoforms of tau, 3R亚型)和Aβ[69]。机制研究发现,AS分泌P-Tau增加,通过线粒体功能障碍损伤AS周围的海马突触和网络结构,小鼠出现空间记忆障碍[35]。从以上的临床、动物和细胞研究中均证实,AS增殖导致了P-Tau增多,进而损害突触和神经元。
7 结 论
综上所述,AS主要是通过能量供给,维持水与电解质平衡、清除或分解突触间神经递质、释放生物活性分子(如IGF-1,或神经胶质递质如谷氨酸、ATP、D-丝氨酸、GABA、牛磺酸等分子)来维持CNS的内稳态。在病理情况下,分泌Aβ和P-Tau、介导神经炎症、促进谷氨酸等释放增加而重吸收障碍等多途径来改变突触和神经元的功能和结构[38],进而影响AD的进展(如图1所示)。在疾病早期,AS主要表现为生理功能丧失,减少神经元的能量和物质代谢;随着病变进展,聚集在Aβ周围有神经毒性作用的A1s的增多[8],细胞膜失去转运谷氨酸和钙离子的能力,分泌大量的促炎因子、Aβ和P-Tau等,促进突触功能障碍及神经元的萎缩和死亡,加重AD病变。
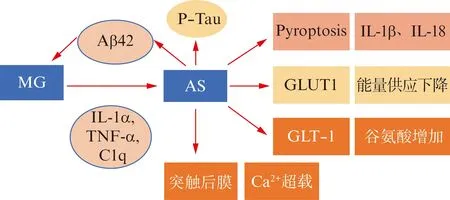
图1 星形胶质细胞在阿尔茨海默病病变过程中的作用模式图
猜你喜欢
自然杂志(2021年6期)2021-12-23 08:24:46
神经损伤与功能重建(2020年11期)2020-12-01 05:01:54
现代装饰(2018年5期)2018-05-26 09:09:01
三门峡职业技术学院学报(2017年1期)2017-06-05 10:17:30
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
中国比较医学杂志(2017年5期)2017-01-17 06:17:05
湖南中医药大学学报(2016年1期)2016-12-01 04:08:21
磁共振成像(2015年1期)2015-12-23 08:52:21
电源技术(2015年5期)2015-08-22 11:18:38
医学研究杂志(2015年12期)2015-06-10 06:57:46
