
CD4-CD8-TCRγδ+ T细胞大颗粒淋巴细胞白血病合并纯红细胞再生障碍性贫血1例并文献复习
2023-10-25 10:15刘金立张玉杰徐腾飞
国际医药卫生导报 2023年20期
刘金立 张玉杰 徐腾飞
1威海市立医院检验科,威海 264200;2威海市立医院病理科,威海 264200
大颗粒淋巴细胞(large granular lymphocyte,LGL)直径约11 μm,较T、B淋巴细胞体积略大,胞质丰富,含较多散在的溶酶体,寿命约数周[1]。LGL 可分为杀伤细胞、自然杀伤细胞两种。LGL 异常克隆性增殖导致大颗粒淋巴细胞白血病(large granular lymphocyte leukemia, LGLL)。LGLL 分为T 细胞大颗粒淋巴细胞白血病(T-cell large granular lymphocyte leukemia,T-LGLL,约占85%)、慢性NK 细胞增殖性疾病及侵袭性NK 细胞白血病[2]。根据T 细胞受体不同,T-LGLL 分为αβ 型T-LGLL 和γδ 型T-LGLL,临床上以αβ 型T-LGLL 多见,γδ 型T-LGLL 少见,T-LGLL 免疫表型多为CD8、CD16、CD57 阳性,仅极少数CD4 阳性或CD4、CD8 双阴性表型[2-3]。本文回顾性分析威海市立医院收治的1例CD4、CD8双阴性γδ型T-LGLL 伴纯红细胞再生障碍性贫血(pure red cell aplasia,PRCA)患者的临床资料并进行文献复习,现报道如下。
临床资料
患者,女,26 岁,1 个月前头晕不适,活动耐量减低,正常体力活动后出现心悸、胸闷、气短症状,遂来威海市立医院就诊。体格检查示:体温38.5℃,颈部、腋窝及腹股沟淋巴结肿大,无畏寒、寒战、咳嗽、咳痰,无出血、黑便、尿色加深、皮疹。2021年3月29日入院后查血常规示:白细胞计数5.9×109/L,中性粒细胞计数0.74×109/L,淋巴细胞计数4.9×109/L,红细胞计数1.07×1012/L,血红蛋白44 g/L,网织红细胞绝对值0.011 3×1012/L,血小板计数473×109/L;生化:肌酐35.8 μmo1/L,尿酸 133.4 μmo1/L,血清铁43.83 μmol/L,不饱和铁结合力4.16 μmo1/L,促红细胞生成素>728.00 mIU/ml;凝血功能:部分凝血活酶时间23 s,纤维蛋白原1.76 g/L;补体C40.19 g/L,β2微球蛋白4.26 mg/L,铁蛋白571.5 μg/L,可溶性转铁蛋白受体0.46 mg/L,转铁蛋白2.1 g/L;直接抗人球蛋白试验阴性,溶血、抗核抗体谱未见异常。心脏B 超示:二尖瓣、三尖瓣少量返流,肺动脉收缩压估测值偏高;腹部B超示:肝胆胰脾未见明显异常;浅表淋巴结B超示:双侧颈部、颌下、腹股沟、腋窝淋巴结肿大,锁骨上淋巴结未见肿大。胸部CT:右肺中下叶少许慢性炎症,心包少量积液。骨穿结果提示:粒、巨二系增生伴红系缺如骨髓象,外周血可见大颗粒淋巴细胞(图1)。流式 TCRVβ检测αβT淋巴细胞未见异常单克隆增生,CD3briCD5dimCD7dimCD4-CD8-γδT细胞占淋巴细胞43.97%(占有核细胞22.57%),TCRγδ1、TCRγδ2 均阴性(图2)。TCRγ、TCRβ 基因重排阳性,IGH、IGK基因重排阴性。基因筛查:STAT3突变4%。骨髓活检:T淋巴细胞轻度增多。染色体检核型:46,XX[20]。
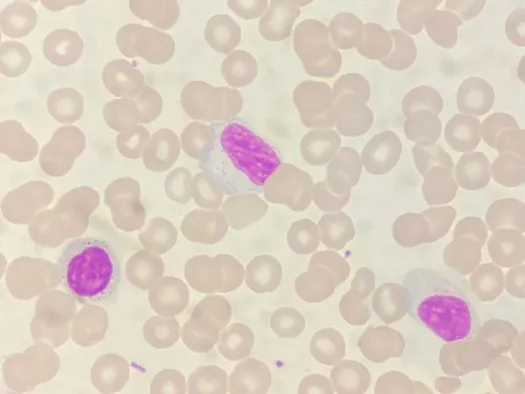
图1 1 例CD4-CD8-TCRγδ+ T 细胞大颗粒淋巴细胞白血病合并纯红细胞再生障碍性贫血患者细胞形态学检查结果(瑞士吉姆萨染色 ×100)

图2 1例CD4-CD8-TCRγδ+ T细胞大颗粒淋巴细胞白血病合并纯红细胞再生障碍性贫血患者流式细胞学检查结果。A为γδ+ T细胞比例增高,B为γδ+ T细胞部分表达CD57,C为异常γδ+ T细胞不表达TCRVδ1和Vδ2
综合以上检查诊断为:CD4-CD8-γδ 型T-LGLL 合并PRCA、肺炎。予抗感染、输血,皮下注射促红细胞生成素、粒细胞集落刺激因子、甲泼尼龙28 mg/d 静滴,联合环抱素125 ng/bid口服治疗3周,复查血常规未见好转,仍需输血支持治疗。遂建议患者抗淋巴细胞球蛋白(antilymphocyte globulin,ALG)或环磷酰胺(cyclophosphamide, CTX)静脉治疗,向患者及其家属交代病情及预后,自愿选择ALG免疫抑制治疗。2021 年4 月15 日起予ALG 治疗,具体方案为:ALG 1.0 g d1~d4、1.25 g d5,过程顺利,输毕出现剑突下疼痛伴发热皮疹、关节肌内酸痛等血清病反应,予糖皮质激素治疗后好转;后患者口服环孢素及曲安西龙联合治疗,血常规提示网织红细胞呈升高趋势,输血间期延长,病情好转出院。
讨论
T-LGLL 是细胞毒性T 淋巴细胞异常克隆性增殖所致的一种少见的惰性淋巴细胞增殖性疾病,占2%~3%[4]。病因及病理机制尚不明确,目前普遍认为由慢性、持续的抗原免疫刺激所引起。在感染暴露或抗原刺激过程中,LGL 短时间内可迅速增殖数万倍,机体在抗原清除后,系统激活选择性消除、诱导LGL 死亡;然而T-LGLL 患者中,诱导细胞凋亡过程功能障碍或被耐受,活化的杀伤性细胞不能有效凋亡,导致外周血LGL 数量升高[5]。另一方面,T-LGLL 患者体内多种细胞信号途径如JAK2/STAT3、鞘脂信号通路、RAS/MEK/ERK 和SFK/PI3K/Akt等被激活,促使LGL 细胞的长期存活[6-7]。研究发现IL-15 和PDGF 是重要的生存信号开关,对LGL 的生存调节有深远影响,可为今后T-LGLL 的诊断提供检测方向[8]。
T细胞发育过程中,CD3+CD4-CD8-是典型的未成熟T细胞表型,多存在于胸腺中。健康个体的外周血中,此表型可见于γδ T细胞或少量αβ T细胞。γδ T细胞根据其V(可变)区分为Vδ1+和Vδ2+,正常γδ T 细胞为多克隆。γδ T 细胞的生理和病理作用尚不完全清楚,但已有研究表明,它们能识别由寄生虫、细菌(尤其是分枝杆菌)刺激机体表达的高度保守的应激蛋白和热休克蛋白[9-10]。目前,已发现γδ T 细胞在自身免疫性疾病如系统性红斑狼疮和感染性疾病中明显升高[11]。
LGLL 临床表现为惰性病程,患者多为老年人,约2/3患者会出现临床症状。主要包括中性粒细胞减少症、贫血和类风湿性关节炎(rheumatoid arthritis,RA)。T-LGLL 患者中性粒细胞减少常见,且会出现复发性细菌感染。贫血主要是自身免疫性溶血性贫血(AIHA)和PRCA。RA 在每一系列的LGLL 患者中都有报道,并且其诊断早于LGLL。5%~35%的患者可能出现输血依赖,20%~50%的患者出现脾肿大,20%血小板减少[12]。研究发现,CD4-CD8-γδ T-LGLL 病例显示出与CD8+γδ T-LGLL 和αβ T-LGLL 相似的临床特征[13]。但外周血大颗粒细胞绝对值两者差异显著:绝大多数αβ 和CD8+γ/δ T-LGLL 病例中,LGL 数量大于0.4×109/L,而4-8-γδ T-LGLL 中LGL 数量一般在正常范围内[14]。本例患者为年轻女性,外周血白细胞计数不高,中性粒细胞计数减低,合并PRCA 但无自身免疫性疾病。文献报道,国内T-LGLL 患者继发PRCA 发生率较高,自身免疫性疾病发生率较低[14];而国外研究报道相反,可能与种族差异有关[15]。T-LGLL 合并PRCA 发病原因与基因缺陷、免疫介导、病毒感染等因素有关,其中CD8+细胞毒T 细胞的克隆性增殖是PRCA 的主要诱因。也有研究推测LGLL 以组织相容性复合物(MHC)限制或非MHC 限制的细胞毒损伤机制导致PRCA 发生[16]。骨髓检查对PRCA 诊断价值较大,但对于T-LGLL作用有限,需要流式细胞术检测辅助诊断。
γδ T 细胞增多最常见的疾病包括三类:γδ T-LGLL、反应性γδ T 淋巴细胞增多症和肝脾T 细胞淋巴瘤(hepatosplenic T cell lymphoma,HSTL)。γδ T-LGLL 与反应性γδ T 淋巴细胞增多症的区别在于:后者T 细胞为多克隆,多表达CD5、CD7、Vδ2,且γδ T淋巴细胞绝对计数<1.8×109/L,淋巴细胞较小,核不规则,不可见核仁或嗜蓝质颗粒。γδ T-LGLL 和HSTL 的区分存在一定的困难;前者多是惰性病程,外周血中可见异常细胞,HSTL 多为侵袭性进程,预后差,很少侵犯外周血[17]。γδ T-LGLL 可表达CD57,同时外周血≥30%的淋巴细胞为典型的大颗粒淋巴细胞、“转化”的免疫母细胞样细胞,胞浆嗜碱性,明显的嗜蓝质颗粒,可见核仁,核深染、圆形或椭圆形;检测到γδT 细胞受体单克隆表达,多表达Vδ1。本病例Vδ1 和Vδ2 均不表达,免疫表型异常。免疫表型上,γδ T-LGLL与HSTL均表现为CD4-/CD8-,但前者常CD57+、TIA-1+、颗粒酶B 阳性,后者CD57-、TIA-1+、颗粒酶B 阴性;HSTL 中的淋巴细胞一致表达CD56[18]。本病例综合上述特点可排除HSTL。
T-LGLL 虽为惰性疾病,但大多数患者最终需要治疗。由于缺乏大规模前瞻性试验,目前无LGLL 标准的治疗方案;有些患者的治疗方法无效,因此对于新治疗方法的需求变得更加迫切。深入探究LGLL 的发病机制可为新的治疗方案提供指导。LGLL 的治疗基于免疫抑制疗法,主要使用甲氨蝶呤(methotrexate,MTX)、CTX、环孢素A(cyclosporine A, CsA)等,糖皮质激素作为辅助治疗[19]。目前,对病程缓慢的无症状LGLL患者,采用等待、观察的方法,可单次注射G-CSF,监测髓系原始细胞增殖分化情况;当患者出现中性粒细胞减少且伴发热时,前期注射G-CSF反应良好者,可以紧急再次给药。对于有症状的患者,免疫抑制治疗的指征包括中度或重度贫血和中度或重度中性粒细胞减少症及合并RA。低剂量MTX 最初用于伴有中性粒细胞减少和/或RA 的T-LGLL 患者。CTX 是T-LGLL 合并贫血患者的首选。Sanikommu 等[20]报道了204 例接受CSA、MTX、CTX 治疗的LGLL 合并PRCA 患者,CTX 的初始有效率最高,反应时间最短。在非典型CD57 阴性T-LGLL 病例中,CTX 的作用尤为显著。CTX 在T-LGLL 合并PRCA 中的作用机制尚不清楚。CTX 减少了直接破坏抗体结合的红母细胞的细胞毒性T淋巴细胞数量,减少了细胞毒性T淋巴细胞介导的造血祖细胞损伤。治疗开始4 个月后,通过血液计数评估是否可达到完全缓解,不能达到部分缓解的患者,则需改用其他药物治疗。MTX、环孢素或CTX与强的松联合使用,治疗效果较好,可导致血液系统快速改善,可能是与正常γδ T细胞中CD5-亚群比CD5+亚群有更高的细胞毒活性有关。
本患者首先采用CsA+甲泼尼龙方案治疗,效果不佳;后选择ALG+环孢素+曲安西龙联合治疗,病情好转。嘌呤类似物的治疗经验较少,但总体有效率较高。考虑到治疗周期短、反应率高、毒性轻、诱导持久缓解等优点,可以考虑使用嘌呤类似物[21]。Tipifarnib是一种法尼酰基转移酶抑制剂,可抑制RAS介导的信号转导,促进LGL的正常凋亡。一项针对LGLL患者的二期临床研究显示:患者骨髓和血液中的LGL 数量减少,使骨髓造血改善,体外造血集落生长增加,还可改善LGLL 患者的肺动脉高压症状[22-23]。CD52 常在白血病T-LGL 上表达,可应用单克隆抗体药物选择性杀死表达CD52 的LGL。阿仑珠单抗是一种人源化的抗CD52 单抗,在一项研究中,阿仑珠单抗的总有效率为50%[24]。目前,阿仑珠单抗正在进行治疗T-LGLL的二期临床试验。
作者贡献声明刘金立:论文撰写与构思;张玉杰:指导、支持性贡献;徐腾飞:对文章的知识性内容作批判性审阅、指导
猜你喜欢
科学大众(2021年6期)2022-01-01
中国民间疗法(2021年14期)2021-08-30
基层中医药(2021年4期)2021-07-22
基层中医药(2021年4期)2021-07-22
当代陕西(2019年23期)2020-01-06
中国科技信息(2015年2期)2015-11-16
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年21期)2015-03-01
郑州大学学报(医学版)(2015年1期)2015-02-27
现代检验医学杂志(2015年5期)2015-02-06
