
QuEChERS-气相色谱-三重四极杆串联质谱法高通量测定畜肉中16种镇静剂药物残留
2023-10-17 09:26:26周颖成昕宁军沈习习王玮
南京农业大学学报 2023年5期
周颖,成昕,宁军,沈习习,王玮*
(1.南京农业大学食品科学技术学院/国家肉品质量安全控制工程技术研究中心/农业农村部肉及肉制品质量监督检验测试中心(南京),江苏 南京 210095;2.农业农村部农产品质量安全中心,北京 100020;温氏食品集团股份有限公司,广东 云浮 527400)
镇静剂类药物有时会被部分养殖户滥用或非法用于动物高密度养殖、长途运输等,以促生长或缓解运输应激[1-2]。然而,该类药物易残留蓄积于动物体内,通过食物链进入人体后,会对人体产生一系列毒副作用,如头晕嗜睡、呼吸抑制以及影响人体中枢神经系统等[3-5]。因此,美国、欧盟、国际食品法典委员会(CAC)等国家和组织均对镇静剂的检出和最高残留限量(maximum residue limits,MRL)做出了相关规定[6]。2020年,《中华人民共和国农业农村部公告 第250号》[7]中关于食品动物中禁止使用的药品及其他化合物清单中也明确禁止使用安眠酮;《食品安全国家标准 食品中兽药最大残留限量:GB 31650—2019》规定阿扎哌隆在猪肉中的MRL为60 μg·kg-1;氯丙嗪和地西泮仅允许使用治疗剂量,但在动物源性食品中不得检出[8]。我国现行有效的动物源性食品中镇静剂类药物残留检测标准相对较少,缺少对巴比妥类、吩噻嗪类及苯二氮卓类等多药物同时检出的方法,且样品前处理也较为繁琐、耗时。因此,建立准确高效且可同时检测畜肉中多种镇静剂类药物残留的方法就显得尤为迫切和重要。
畜肉基质较为复杂,筛选合适的样品前处理方法有助于提高检测灵敏度,减少基质干扰。目前,动物性食品中镇静剂类药物残留检测的前处理方法主要有液-液萃取(liquid-liquid extraction,LLE)[9]、固相萃取(solid-phase extraction,SPE)[10]、固相微萃取(solid phase micro extraction,SPME)[11]、QuEChERS(quick,easy,cheap,effective,rugged,safe)[12]等,前3种前处理方法虽可有效分离药残组分,但操作繁琐耗时,定量检测精确度低。QuEChERS方法具有操作简单、快速高效、溶剂消耗量少及回收率高等优点,现已广泛应用于食品、生物、医药等领域[13-14]。已有的镇静剂类药物残留检测的方法主要有酶联免疫法(enzyme-linked immunosorbent assay,ELISA)[15]、高效液相色谱法(high performance liquid chromatography,HPLC)[16]、气相色谱-质谱联用法(gas chromatography-mass spectrometry,GC-MS)[17]、液相色谱-串联质谱法(liquid chromatography-tandem mass spectrometry,LC-MS/MS)[18-19]、气相色谱-三重四极杆串联质谱法(gas chromatography-triple quadrupole tandem mass spectrometry,GC-MS/MS)[20]等,其中LC-MS/MS和GC-MS/MS因其灵敏度高并可同时检测多种化合物,已逐渐成为检测多药物残留的主要方法[21]。与LC-MS/MS法相比,GC-MS/MS法因具有更好的抗干扰能力,分析结果准确可靠,适用于痕量物质的检测,且仪器成本低,操作简单易于普及[22]。已有研究建立的GC-MS/MS法主要聚焦于检测血浆和尿液基质中的镇静剂类药物[23-24],较少用于检测动物性食品。因此,本研究基于QuEChERS前处理方法,构建一种可同时检测畜肉中16种镇静剂类药物的GC-MS/MS法,旨在为我国动物性食品中镇静剂类药物残留的高通量快速检测以及食品安全监管提供技术支持和保障。
1 材料与方法
1.1 材料与试剂
生鲜猪肉、牛肉、羊肉购于江苏省南京市农贸市场;阴性畜肉样品由农业农村部肉及肉制品质量监督检验测试中心(南京)提供。
标准品溶液:巴比妥、苯巴比妥、安眠酮、地西泮、氯丙嗪、氯氮卓、咪达唑仑、三氟拉嗪盐酸盐、阿扎哌隆、硝西泮、丙酰二甲氨基丙吩噻嗪盐酸盐、氯硝西泮、氯氮平、艾司唑仑、阿普唑仑、三唑仑(质量浓度均为100 μg·mL-1)均购于天津阿尔塔科技有限公司。
试剂:乙腈、甲醇、氨水、乙酸乙酯、异丙醇(均为色谱纯)购于上海安谱实验科技股份有限公司;无水硫酸钠(分析纯)购于西陇化工股份有限公司;氯化钠(分析纯)购于广东光华科技股份有限公司;柠檬酸三钠(C6H5Na3O7,分析纯)购于上海源叶生物科技有限公司;柠檬酸二钠倍半水合物(C6H6Na2O7·1.5H2O,分析纯)购于德国CNW科技公司;十八烷基键合硅胶(C18)、N-丙基乙二胺(PSA)购于德国CNW科技公司,粒径均为40 μm;试验用水均为一级水。
1.2 仪器与设备
TSQ 8000 Evo三重四极杆气质联用仪、TR-Pesticide Ⅱ毛细管色谱柱(30 m×0.25 mm×0.25 μm),购于美国Thermo Fisher Scientific公司;arium®advance EDI纯水仪、D-16C高速冷冻离心机、SECURA313-1CN电子天平购于德国Sartorious公司;Vortex Genius漩涡混匀器购于德国IKA公司;N-EVAP型氮吹仪购于美国Organomation公司;PTFE针头过滤器(孔径0.22 μm)购于南通迈博瑞生物膜技术有限公司。
1.3 试验方法
1.3.1 标准溶液的配制1)混合标准中间溶液(5 μg·mL-1):分别精确移取16种镇静剂标准品各0.5 mL于10 mL容量瓶中,用乙酸乙酯稀释定容至10 mL,配制成质量浓度为5 μg·mL-1的混合标准中间溶液,-20 ℃避光保存,有效期30 d。
2)混合标准工作溶液(0.5 μg·mL-1):精确移取标准中间溶液0.1 mL于1 mL容量瓶中,用乙酸乙酯稀释定容至1 mL,配制成质量浓度为0.5 μg·mL-1的标准工作溶液,-20 ℃避光保存,有效期7 d。
3)纯溶剂混合标准曲线工作溶液:精确量取0.5 μg·mL-1的混合标准工作液适量,乙酸乙酯稀释,分别配制质量浓度为1.0、5.0、10.0、20.0、50.0、100.0、200.0 ng·mL-1的系列混合标准工作溶液,现配现用。
4)基质匹配混合标准工作溶液:精确量取0.5 μg·mL-1的混合标准工作液适量,分别添加至经提取、净化步骤处理的7份空白试料复溶液中,配制质量浓度为1.0、5.0、10.0、20.0、50.0、100.0、200.0 ng·mL-1的系列基质匹配混合标准工作溶液,现配现用。
1.3.2 样品前处理1)提取:准确称取5.00 g均质样品于50 mL离心管中,加入10 mL提取剂,再加入一定量缓冲盐包,振荡涡旋2 min,4 ℃、10 000 r·min-1离心5 min。
2)净化:转移上清液于装有一定量吸附剂的50 mL离心管中,涡旋混匀1 min,4 ℃、10 000 r·min-1离心5 min;再次转移上清液于新的50 mL离心管中,在35~40 ℃下氮吹近干,加入1 mL乙酸乙酯复溶,振荡涡旋1 min;取复溶液,经0.22 μm PTFE针头过滤器过滤后供GC-MS/MS测定使用。
1.3.3 样品前处理条件优化1)提取剂的优化:均质样品中添加20 μg·kg-1混合标准溶液,按照1.3.2节方法进行样品前处理,选用如下5种提取剂分别提取并分析不同提取剂的回收率:乙腈、1%氨水乙腈、乙酸乙酯、乙酸乙酯-异丙醇(5∶1,体积比)、甲醇。
2)缓冲盐包的优化:使用最佳提取试剂,按照1.3.2节方法进行样品前处理,分析4种缓冲盐包的萃取效果,4种缓冲盐包为:缓冲盐包A(2 g Na2SO4+1 g NaCl)、缓冲盐包B(1 g C6H5Na3O7+0.5 g C6H6Na2O7·1.5H2O+2 g Na2SO4)、缓冲盐包C(1 g C6H5Na3O7+0.5 g C6H6Na2O7·1.5H2O+2 g Na2SO4+1 g NaCl)、缓冲盐包D(1 g C6H5Na3O7+0.5 g C6H6Na2O7·1.5H2O+4 g Na2SO4+1 g NaCl)。
3)吸附剂用量的优化:在最佳提取条件下,按照1.3.2节方法进行样品前处理,对PSA、C18及Na2SO4的用量进行优化。选取5种吸附剂组合,分别为吸附剂组合A(2 g Na2SO4+0.2 g PSA+0.2 g C18)、吸附剂组合B(2 g Na2SO4+0.15 g PSA+0.1 g C18)、吸附剂组合C(2 g Na2SO4+0.1 g PSA+0.15 g C18)、吸附剂组合D(2 g Na2SO4+0.1 g PSA+0.05 g C18)、吸附剂组合E(2 g Na2SO4+0.1 g PSA),分析不同吸附剂用量的净化效果。
1.3.4 GC-MS/MS检测条件优化1)气相色谱条件优化。色谱柱:TR-Pesticide Ⅱ毛细管色谱柱(30 m×0.25 mm×0.25 μm);载气:高纯氦气(99.999%);恒流模式流速:1.0 mL·min-1;进样方式:不分流进样;进样量:1 μL;进样口温度:300 ℃。将5 μg·mL-1的混合标准溶液注入仪器进样口端,选取3种升温程序方案:①初始温度70 ℃,以50 ℃·min-1升温至250 ℃,保持1 min,再以5 ℃·min-1升温至300 ℃,保持5 min;②初始温度70 ℃,以35 ℃·min-1升至200 ℃,再以5 ℃·min-1升至300 ℃,保持5 min;③初始温度70 ℃,以50 ℃·min-1升至200 ℃,再以10 ℃·min-1升至300 ℃,保持5 min。通过分析16种药物的分离效果,确定最佳升温程序。
2)质谱条件优化。离子源:电子轰击离子源(EI);电离能量:70 eV;离子源温度:300 ℃;离子传输线温度:280 ℃;溶剂延迟时间:4 min;扫描离子范围:40~550m/z;扫描方式:多反应监测模式(multiple reaction monitor,MRM)。在上述条件下,本试验通过仪器AutoMRM功能模块进行母离子、子离子及碰撞能量的优化。
1.4 方法学验证
1.4.1 基质效应(matrix effect,ME)ME是指样品中其他成分对目标化合物质量浓度的影响,即基质对分析方法准确性的干扰[25]。以基质匹配标准曲线斜率(k1)与纯溶剂标准曲线斜率(k2)相对比值评价基质效应,即ME=(k1/k2-1)×100%[26]。当|ME|在0%~25%时,说明基质对待测物具有弱基质效应,即基质效应不显著;当|ME|在25%~50%时,说明基质对待测物具有中等强度的基质效应;当|ME|大于50%时,则说明基质对待测物具有较强基质效应,基质效应较显著[27]。
1.4.2 线性范围、检出限及定量限的确定基于已建立的GC-MS/MS法,分别以阴性猪肉、牛肉、羊肉为基质,配制7个不同梯度质量浓度(1~200 ng·mL-1)的混合基质匹配标准工作溶液。以定量离子质量色谱峰面积为纵坐标(Y),对应溶液质量浓度(ng·mL-1)为横坐标(X),绘制标准曲线,求得回归方程,计算平方相关系数(R2)。分别在阴性畜肉样品中添加1 ng·mL-1混合标准溶液进行测定,以信噪比(S/N)=3计算各化合物的检出限(limit of detection,LOD),以S/N=10计算各化合物的定量限(limit of quantitation,LOQ)[28]。
1.4.3 准确度和精密度试验依据《实验室质量控制规范 食品理化检测:GB/T 27404—2008》,在阴性畜肉中分别添加3个水平(LOQ、2倍LOQ、10倍LOQ)的混合标准品溶液,进行加标回收试验,每个水平平行测6次,进行3次批间重复。计算平均回收率、批内相对标准偏差(relative standard deviation,RSD)和批间RSD。
1.4.4 方法的实际应用选用市售生鲜肉20份,其中猪肉10份,牛肉、羊肉各5份。参照1.3.2节和1.3.3节分别对样品进行前处理和上机测定,以验证该方法的实际应用效果。
2 结果与分析
2.1 样品前处理条件优化
2.1.1 提取试剂的优化由图1可知:乙腈在猪肉、牛肉和羊肉3种基质中均具有最佳提取效果,其回收率在60%~120%的目标化合物数量最多。因此,本试验选用乙腈作为提取溶剂。
2.1.2 缓冲盐包的优化由图2可知:缓冲盐包C在3种基质中均呈现最佳的萃取效果,其回收率在60%~120%的目标化合物数量最多。因此,本试验选用缓冲盐包C(1 g C6H5Na3O7+0.5 g C6H6Na2O7·1.5H2O+2 g Na2SO4+1 g NaCl)作为缓冲盐包。
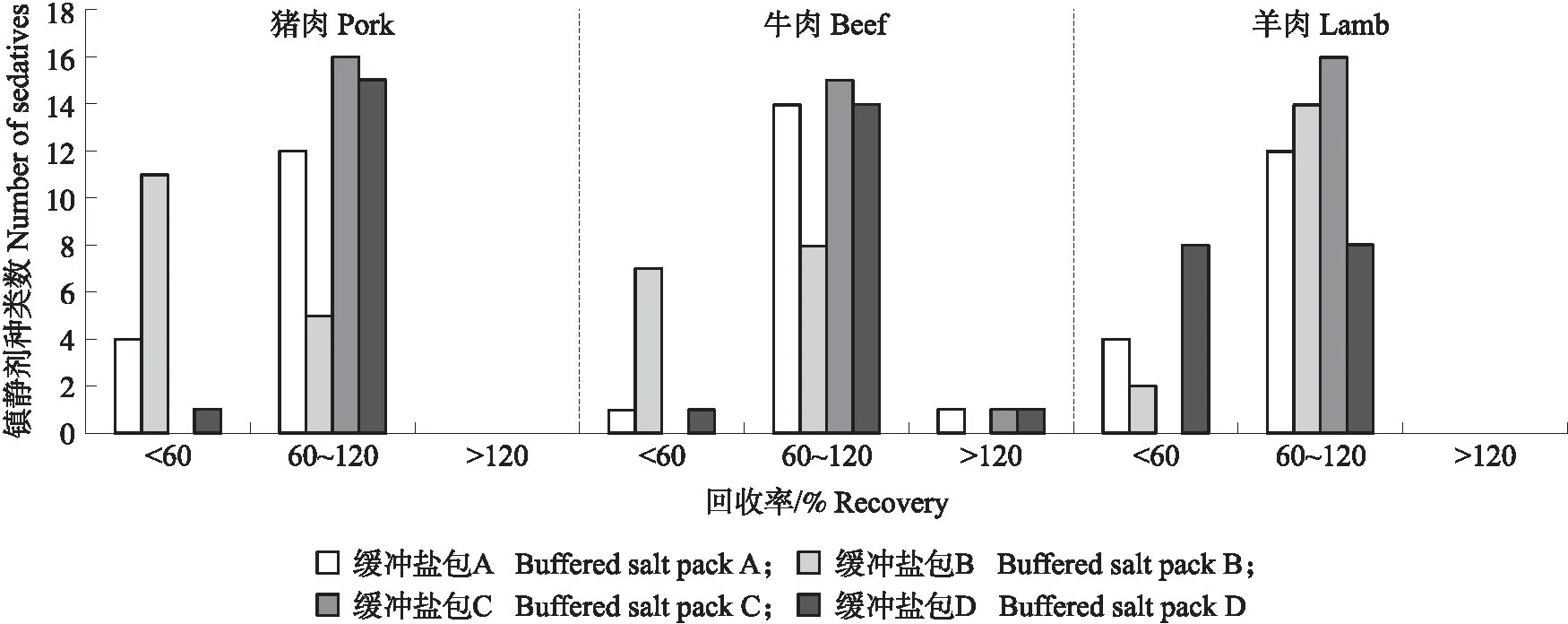
图2 不同缓冲盐包对16种镇静剂药物回收率的影响Fig.2 Effect of different buffered salt packs on the recovery of 16 sedative drugs
2.1.3 吸附剂用量的优化由图3可知:吸附剂组合E在3种畜肉基质中均具有最佳净化效果,其所有目标化合物的回收率均为60%~120%。因此,本试验选用吸附剂组合E(2 g Na2SO4+0.1 g PSA)作为净化吸附剂。
2.2 仪器条件优化
2.2.1 气相色谱条件的确定由图4可知:当升温程序为方案①时,16种镇静剂药物分离效果好,响应高,无杂峰,可实现定性分析。
2.2.2 质谱条件的确定本试验在1.3.3节优化的色谱条件下,将5 μg·mL-1的混合标准溶液以单针进样方式注入GC-MS/MS中进行全扫描,选择强度高的碎片离子作为母离子,再对母离子进行选择反应监测模式扫描,选择2个强度高且稳定的碎片离子作为子离子,然后通过子离子优化碰撞能量,最终确定16种镇静剂药物的质谱参数(表1)。按照上述条件,获得16种镇静剂类药物混合标准溶液的多反应监测(MRM)色谱图,如图5所示。

表1 16种镇静剂药物的保留时间、定性离子对、定量离子对及碰撞能量Table 1 Retention time,qualitative ion pair,quantitative ion pair and collision energy of 16 sedative drugs

图5 16种镇静剂药物的多反应监测(MRM)色谱图Fig.5 Multiple reaction monitor chromatograms of 16 sedative drugs
2.3 基质效应评价
由表2可知:巴比妥、苯巴比妥、安眠酮、地西泮以及硝西泮在3种畜肉基质中均呈现出基质增强效应,而其他物质则表现为基质抑制效应。其中,硝西泮在猪肉、牛肉、羊肉3种基质中的|ME|分别为6.89%、3.46%、6.98%,说明其基质效应不显著。此外,本试验虽通过优化前处理方法降低了基质干扰,但苯巴比妥、安眠酮、氯丙嗪、氯硝西泮、艾司唑仑、阿普唑仑、三唑仑在3种畜肉基质中仍表现为较强的基质效应(|ME|>50%),其中安眠酮的|ME|在牛肉和羊肉基质中均高于160%。因此,为校正基质效应和提高结果准确性,本试验最终采用基质匹配标准曲线法。
2.4 方法的线性范围、检出限和定量限
由表2可知:16种镇静剂药物在1~200 ng·mL-1范围内线性范围良好(R2≥0.992 8),检出限为0.01~0.30 μg·kg-1,定量限为0.10~1.00 μg·kg-1。
2.5 方法的准确度和精密度
由表3可知:16种镇静剂药物的回收率为64.83%~112.04%,批内RSD为1.45%~8.96%,批间RSD为4.84%~14.88%,均符合方法学要求。
2.6 方法实际应用评价
为验证本方法的实际应用效果,分别在批发市场和屠宰场随机选取20份畜肉作为供试材料,采用本试验建立的方法进行检测。由表4可知:在1份猪肉和牛肉中均检出巴比妥(1.58、0.77 μg·kg-1),1份羊肉中检出阿扎哌隆(0.34 μg·kg-1),1份猪肉中检出地西泮(未超过本方法LOQ:0.1 μg·kg-1);其他供试材料均未检出16种镇静剂类药物残留。

表4 市售生鲜畜肉中镇静剂类药物残留的检测结果Table 4 Detection results of sedative drug residues for municipal sold fresh livestock meats μg·kg-1
3 讨论
近年来,食品安全问题日益成为人们关注的焦点和热点,其中动物性食品中兽药残留问题尤为显著。镇静剂类药物为禁用兽药,但在动物养殖过程中违禁使用该类药物的现象仍屡见不鲜。因此,亟需建立动物性食品中痕量镇静剂类药物残留检测方法。本研究基于GC-MS/MS法检测了16种镇静剂药物残留,所有目标化合物无需衍生化就可实现较好的分离度和重现性,并采用QuEChERS法对样品进行前处理,通过优化提取剂、缓冲盐包及吸附剂使用量,显著降低了畜肉中蛋白质、色素、脂肪等基质干扰,有助于提高目标化合物的回收率。此外,与现有样品前处理方法相比[29-30],本方法省略了正己烷除脂和过柱步骤,大大简化并加快了样品前处理过程,提高了检测效率并降低了成本。
本试验对市售生鲜畜肉检测结果显示,待测样中检出巴比妥、地西泮及阿扎哌隆等违禁兽药,此类药物严禁在动物饲料和饮用水中使用,且不得在动物源性食品中检出[1]。究其原因可能是养殖户受利益驱动,在养殖及运输环节非法使用镇静剂类药物,导致该类药物在畜肉中残留超标[31]。
本试验构建了一种QuEChERS结合GC-MS/MS高通量检测畜肉中16种镇静剂药物残留的方法,16种镇静剂类药物的检出限为0.01~0.30 μg·kg-1,定量限为0.10~1.00 μg·kg-1,均低于其他文献报道[32-33]。在0.10~10.00 μg·kg-1的加标水平下,16种药物的平均回收率为64.83%~112.04%,批内RSD为1.45%~8.96%,批间RSD为4.84%~14.88%。该方法的回收率和精密度均符合《实验室质量控制规范食品理化检测:GB/T 27404—2008》对回收率和精密度的要求[34]。该方法可实现市售畜肉中多种镇静剂类药物残留的高通量快速检测,对于我国动物性食品国际贸易及保障消费者健康与安全具有重要意义。
猜你喜欢
中国乡村医药(2023年22期)2023-11-30 06:35:26
肉类研究(2019年11期)2019-12-19 01:59:27
养生保健指南(2019年10期)2019-12-16 01:46:38
福建质量管理(2019年12期)2019-07-01 09:58:12
恋爱婚姻家庭·养生版(2018年12期)2018-01-15 02:25:26
中老年健康(2017年8期)2017-12-16 23:25:36
中西医结合心血管病电子杂志(2017年6期)2017-09-07 21:11:19
中国实用医药(2016年28期)2016-12-07 07:55:18
家庭医药·快乐养生(2015年7期)2015-09-10 07:22:44
发明与创新·中学生(2014年12期)2014-12-16 17:32:09
