
快速热解铂前体合成高分散的Pt/HY催化剂及其萘深度加氢性能
2023-10-07 12:35王兰江梁瑜汤琼唐明兴李学宽刘雷董晋湘
化工进展 2023年8期
王兰江,梁瑜,汤琼,唐明兴,李学宽,刘雷,董晋湘
(1 太原理工大学化学工程与技术学院,化学产品工程山西省重点实验室,山西 太原 030024;2 中国科学院山西煤炭化学研究所,山西 太原 030001)
我国是煤焦油生产的大国,目前国内煤焦油的年产量已超过2650 万吨。焦化萘作为煤焦油中的主要提取物(约占10%)是工业上重要的稠环芳香烃[1],其深度加氢反应的研究意义重大,产物十氢萘广泛用作涂料、树脂、橡胶等的溶剂、航空煤油添加剂、除漆剂、燃料电池储氢媒介等[2-5]。由于萘加氢是可逆的放热反应,就热力学而言,低温有利于该反应[6-7]。传统的非贵金属(Ni、Mo、Co、W)加氢催化剂具备较好的抗硫性,在芳烃加氢中应用较广[8]。但普遍存在催化活性较差、加氢深度低、反应温度及压力高等缺点[9]。与之相比,贵金属催化剂具有较强的活化芳烃分子和解离氢的能力,在较低的温度和压力下便可实现芳烃的深度加氢[10-11]。
由于贵金属地壳丰度较低导致生产成本持续攀升,目前在提高原子利用率节约资源的诉求下,制备高分散的贵金属催化剂已然成为首要选择[12]。传统的浸渍法由于操作简便,常被应用于将金属离子引入几乎所有类型的沸石孔道中[13]。但由于金属离子与孔壁间相互作用力的存在,使其难以被大量的引进孔道,且在溶剂表面张力的影响下,极易聚集沉积于载体表面[14-15]。为此通过物理气相沉积(PVD)将尺寸小且易挥发的金属络合物[Pt(acac)2]均匀地引入孔道的做法开始被应用,然而由于高分散的Pt 物种表面能较高,在传统持续高温煅烧去除配体的过程中极易发生迁移聚集[16-18],因此有必要改进煅烧方法以抑制Pt物种团聚。
本文以乙酰丙酮铂作为前体物理气相沉积到HY 沸石孔道中,通过快速热处理的方式去除有机配体用于制备高度分散的负载型铂基催化剂。快速热处理提供的适度能量仅致力于将Pt(acac)2热解,不再为解离后的Pt 物种提供过多的迁移能量,有效抑制了Pt 物种团聚并将其牢固地锚定在沸石孔道内。基于上述合成方法,制备了高度分散的0.5Pt/HY催化剂,并通过一系列技术对催化剂的结构和性质进行了系统的表征。通过与传统煅烧法所制催化剂对比,揭示了高分散负载型铂基催化剂在萘深度加氢反应中的催化机理。
1 实验部分
1.1 实验试剂
萘(C10H8,≥98%)、正癸烷(C10H22,≥98%)、正辛烷(C8H18,>99%)、乙酰丙酮铂(C10H14O4Pt,98%),上海阿拉丁生化科技股份有限公司;HY(SiO2/Al2O3=5.4),天津南化催化剂有限公司;无水乙醇(C2H6O,≥99.7%),天津市天力化学试剂有限公司。
1.2 催化剂制备
将一定量的Pt(acac)2和HY在50℃的乙醇溶液中搅拌浸渍3h,然后在90℃烘箱中干燥10h。之后,将样品充分研磨装入微孔物理吸附仪的密封管中,并加热到70℃,在压力降至500μmHg(1μmHg=0.13332Pa)后保持1h。之后,升温至90℃保持10h,以去除孔道中的溶剂。最后,将温度升至130℃并保持3h,以获得前体Pt(acac)2@HY。
配体去除过程如图1 所示:称取适量前体Pt(acac)2@HY平铺于瓷舟中并提前放置在管式炉中的冷却区,待管式炉升至450℃后,开大氩气流量(>120mL/min),将样品拉至加热区并计时1min,计时结束后将样品推至冷却区静置10min,使样品在氩气的吹扫下快速降温,最终获得目标产物0.5Pt/HY(0.5代表Pt的理论负载量为0.5%)。为对比传统煅烧法,称取适量前体Pt(acac)2@HY 置于管式炉中以2℃/min的升温速率升至350℃并在该温度下保持1h,获得目标产物0.5Pt/HY-T(T代表传统煅烧)。通过ICP 测得以上催化剂的实际Pt 负载量,具体数据见表1。
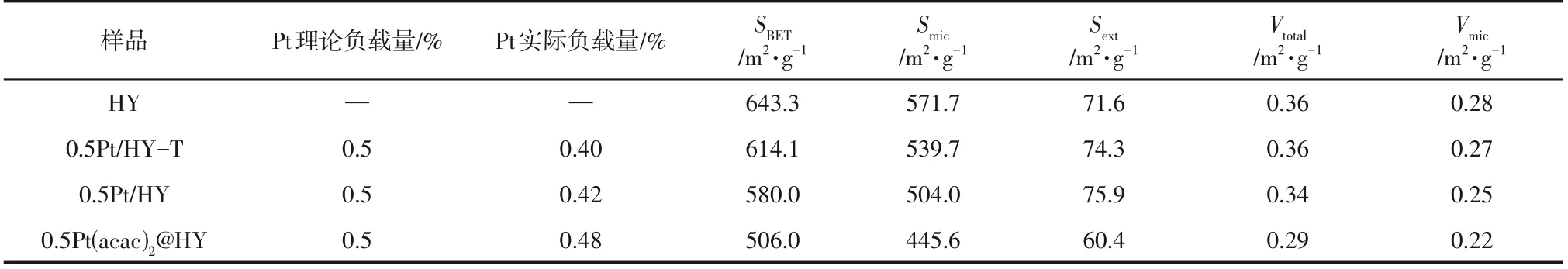
表1 样品的孔结构参数及ICP表征结果
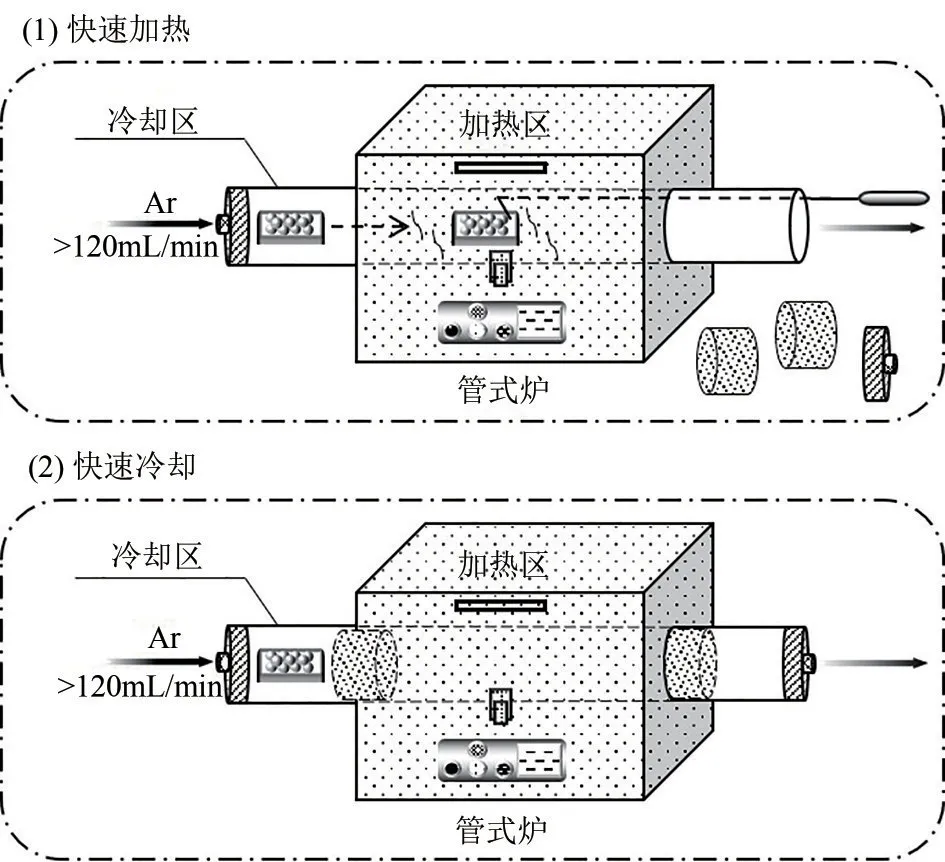
图1 快速热处理法去除有机配体的示意图
1.3 催化剂表征
采用热重分析仪(Labsystems EVO 法国塞塔拉姆仪器公司)对样品中配体去除情况进行了分析,载气选用氩气(20mL/min);催化剂的孔结构参数使用分析仪(ASAP2000,麦克默瑞提克(上海)仪器有限公司)通过氮吸附在77.4K下测定,测量前催化剂样品在130℃下脱气10h;使用日本理学Rigaku MiniflexⅡ型X射线衍射仪(XRD),对样品的物相结构进行确认,CuKα(λ=0.15418nm);程序升温脱附(H2-TPD)、程序升温还原(H2-TPR)均在麦克Auto ChemⅡ2920 化学吸附仪中进行测定,用热导检测器(TCD)检测并记录谱图;原位漫反射红外傅里叶变换(CO-DRIFT)实验在配有Harrick Scientific DRIFTS 池的Bruker INVIO R 光谱仪上进行;使用200kV 的加速电压(JEM-2100F)进行高分辨率透射电子显微镜(HRTEM);采用XPS(Axis Ultra DLD,Kratos)分析样品表面Pt 物种的价态;采用化学吸附仪(麦克Auto ChemⅡ2920)进行CO 脉冲化学吸附实验,根据CO 的吸附量,基于CO/Pt 的比值为2,计算表面Pt 的原子数。
1.4 催化剂活性测试
萘加氢反应的催化性能评估均在25mL 不锈钢高压釜(内含聚四氟乙烯内衬)中进行。搅拌速率设置为500r/min以消除传质影响。选用正癸烷作为溶剂、正辛烷作为内标物配置出0.05mol/L 的萘溶液。反应前向上述反应器中加入5mL 反应液、20mg 催化剂,并用高纯H2吹扫5 次后通入H2加压至3MPa。在气相色谱仪(Shimadzu 2010 pro、FID检测器和DB-5-HT 柱)上对产物进行定量分析。转化率、目标产物选择性、产率和TOF(转换频率)用式(1)~式(4)计算。
2 结果与讨论
2.1 合成策略及配体去除
在合成高分散Pt 基催化剂的过程中,采用PVD法将大部分的Pt(acac)2均匀地引入到HY沸石的孔道中,传统的持续煅烧虽然有利于去除配体,但极易使活性金属聚集,为此做出改进以期通过快速热处理来抑制解离后Pt 物种的迁移。氩气气氛下通过热重(TG)分析对前体0.5Pt(acac)2@HY的热解温度进行了检测(图2)。结果表明,100℃左右的质量损失归因于水的脱除,Pt(acac)2在氩气气氛下的分解温度较高,直到350℃时失重曲线才趋于平稳,表明传统煅烧法(350℃)和快速热处理法(450℃)均能有效去除配体。
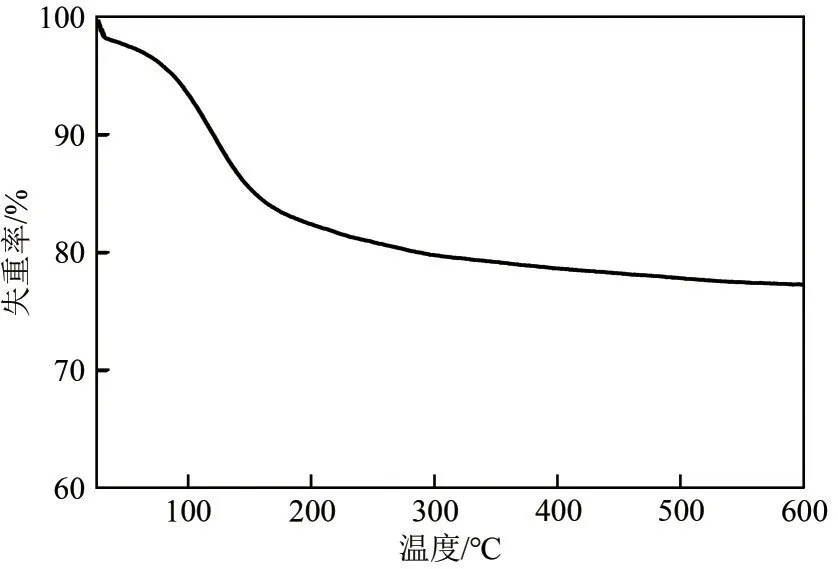
图2 前体0.5Pt(acac)2@HY的热重分析结果
采用N2吸附-脱附表征揭示了系列催化剂的结构特性(图3),样品HY、0.5Pt/HY-T 和0.5Pt/HY的吸附量均在相对压力(p/p0)为0~0.2的范围内急剧增加,呈现出典型的Ⅰ型等温线,且在p/p0为0.4~0.99的范围内,未出现明显的回滞环,表明样品主要存在微孔结构,在制备过程中孔结构没有受到破坏[19]。此外,如表1所示,0.5Pt(acac)2@HY的比表面积(SBET)、微孔体积(Vmic)和微孔面积(Smic)均小于HY、0.5Pt/HY-T 和0.5Pt/HY 样品,表明Pt(acac)2被引入到了HY 的孔道中,在通过传统煅烧和快速热处理去除配体后孔内空间得到释放,这也进一步证明了两种方法在去除配体上的有效性。结合ICP 结果(表1)可以发现在Pt 含量基本相同的条件下(0.5Pt/HY-T 为0.40,0.5Pt/HY 为0.42),0.5Pt/HY-T 具有更大的Smic和Vmic,表明在持续高温影响下0.5Pt/HY-T 上解离后的Pt 物种更倾向于团聚成大颗粒甚至迁移出微孔。与之相反,0.5Pt/HY上的Pt物种高度分散在微孔中,从而占据了更多的孔内空间。
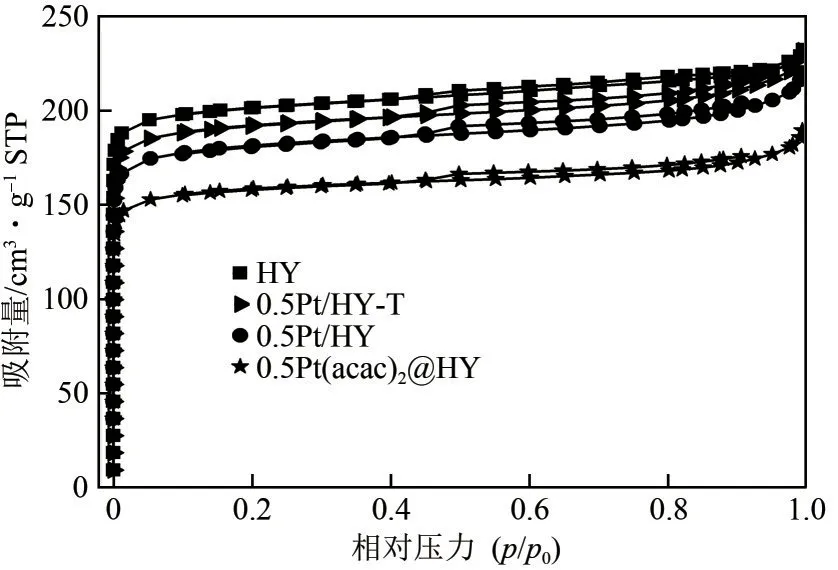
图3 样品的N2吸附-脱附等温线
2.2 催化剂粒度分布及表面特性
0.5Pt/HY和0.5Pt/HY-T的XRD衍射谱图如图4所示,两催化剂在2θ值为15.58°、18.76°、20.49°、23.80°、27.20°、31.56°处观察到了属于HY 的特征衍射峰,未检测到Pt物种的衍射峰[20],这表明在孔道限域的作用下大部分的Pt物种高度分散在HY载体上且在制备过程中载体结构并未被破坏。
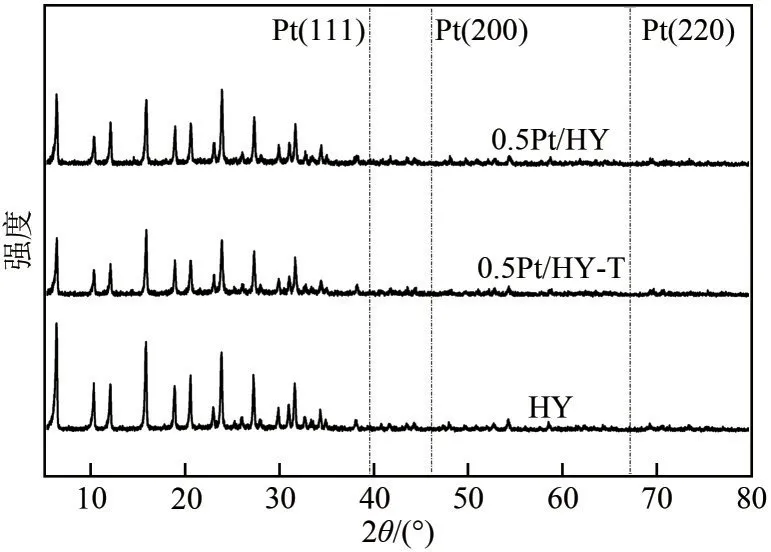
图4 系列样品的粉末XRD谱图
H2还原后的0.5Pt/HY 和0.5Pt/HY-T 催化剂的H2-TPD 表征结果如图5 所示,两催化剂的H2吸附量分别为82.4mmol/g 和49.0mmol/g,扣除纯HY 载体吸附的H2量后,计算结果显示0.5Pt/HY 上表面Pt物种吸附的H2量是0.5Pt/HY-T上的2.4倍,表明0.5Pt/HY具有更高的分散度,可吸附更多的H2,这是能够高效活化H2的必要前提。
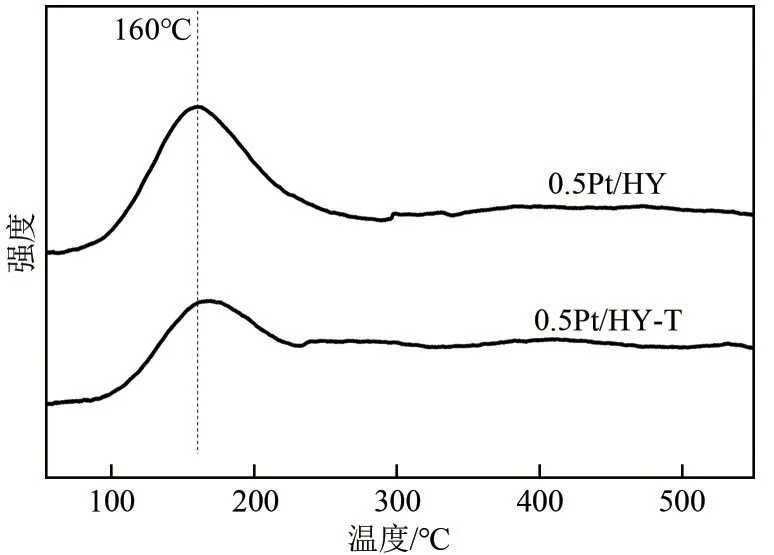
图5 0.5Pt/HY和0.5Pt/HY-T催化剂的H2-TPD谱图
H2-TPD 的表征结果已经证实了Pt 在0.5Pt/HY上的分散度远高于0.5Pt/HY-T,但Pt 物种确切的粒度分布状态依然难以确定。CO-DRIFT光谱是研究Pt/HY催化剂表面性质的有力工具,红外波段位于2085cm-1附近以及1860cm-1附近的振动峰分别归属于CO在Pt团簇上的线性吸附和在Pt纳米粒子上的桥式吸附[9,21],2095cm-1至2200cm-1范围内的振动峰归属于CO在Pt单原子上的线性吸附[包含多羰基物种Ptδ+(CO)n与单羰基物种Ptδ+(CO)][22-23]。如图6所示,由快速热处理法制得的0.5Pt/HY催化剂上单原子与团簇共存,且并未观测到桥式吸附峰,然而在传统煅烧法制得的0.5Pt/HY-T 催化剂上仅观测到了CO在Pt团簇上的线性吸附峰,说明在使用传统煅烧法除配体的过程中,Pt物种会持续地发生迁移并大量团聚,导致Pt物种仅以团簇的形式存在。
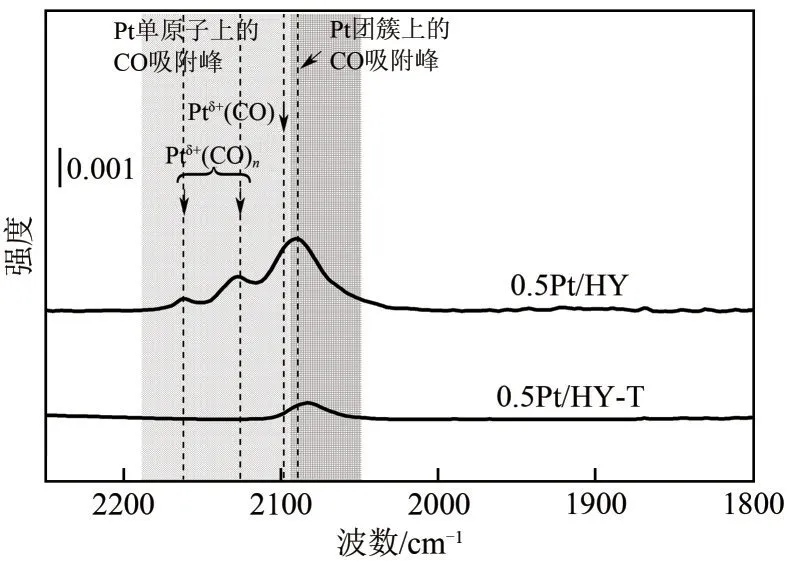
图6 0.5Pt/HY和0.5Pt/HY-T催化剂的原位CO-DRIFT光谱图
采用HRTEM表征进一步考察了Pt物种的粒度分布(图7)。结合CO-DRIFT表征可知0.5Pt/HY上Pt物种以单原子和团簇共存的形式存在,但由于分辨率的限制,HRTEM 难以用于直接观测单个金属原子,因此在0.5Pt/HY 上(Pt 平均粒径d=0.9nm)仅观察到了较小且分布均匀的Pt 团簇,表明快速热处理法可以在配体去除过程中有效抑制Pt 物种的团聚,相反0.5Pt/HY-T催化剂上(Pt平均粒径d=2.4nm)仅能观测到少量的Pt 团簇,其余为分布欠均匀的Pt 纳米粒子且分布于载体的外缘,这表明长时间的高温(350℃)煅烧导致Pt 物种不断迁移并最终大量聚集甚至迁移出孔道。综上,与传统煅烧法相比,快速热处理法采用极速升温后快速降温的策略为前体的热解提供了适度的能量,这有效地避免了解离后Pt 物种的大规模迁移从而利于获得高度分散的Pt物种。

图7 0.5Pt/HY和0.5Pt/HY-T催化剂的HRTEM图
通过XPS 对0.5Pt/HY 和0.5Pt/HY-T 催化剂中Pt的电子价态进行了分析(图8),所有校准均基于284.8eV下C 1s的标准结合能。结合能在71.10eV和74.43eV附近的特征峰被分配给Pt0物种,72.50eV和75.83eV 附近的特征峰分配给Pt2+物种[24],74.65eV附近的特征峰值分配给Al 2p 物种[25]。结果表明,0.5Pt/HY 中Pt0占66.1%,Pt2+占33.9%。而在0.5Pt/HY-T 中,Pt0占84.8%,Pt2+仅占15.2%。由于传统煅烧法升温缓慢且煅烧时间过长,导致解离后的Pt物种有充足的时间去聚集成较大颗粒(颗粒内部主要为Pt0物种),从而具有了较高的Pt0占比,然而0.5Pt/HY 上的Pt 物种高度分散从而具有更多的Pt2+占比,这与HRTEM所观察到的结果一致。
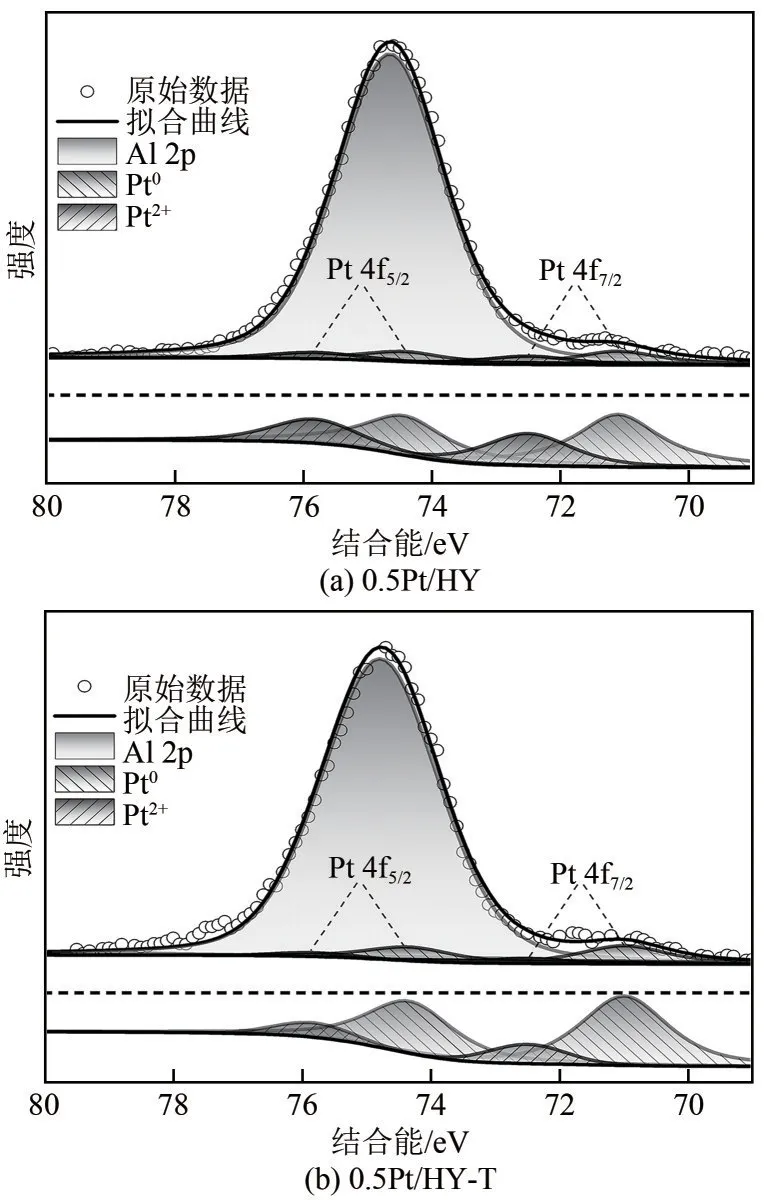
图8 0.5Pt/HY和0.5Pt/HY-T催化剂的Pt 4f XPS谱图
通过H2-TPR 表征对制备的催化剂上Pt物种的还原性能进行考察(图9)。结果显示0.5Pt/HY 在184℃处和435℃处各有一个还原峰,分别归属于易还原Pt物种和难还原Pt物种。与团簇相比,Pt单原子由于具有较高的表面能而更容易被还原,再结合XPS 和CO-DRIFT 分析可推测184℃和435℃处分别为Pt2+单原子和Pt2+团簇的还原峰。此外,0.5Pt/HY-T的还原峰出现在更高温度(450℃),表明传统煅烧法为配体的去除提供了过度的能量,导致解离后的Pt物种不断迁移聚集形成了更稳定的大颗粒[26]。
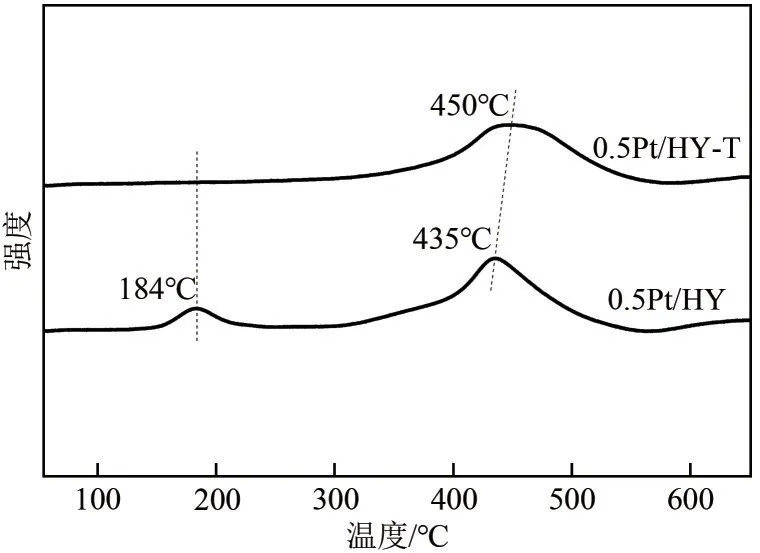
图9 0.5Pt/HY和0.5Pt/HY-T催化剂的H2-TPR实验结果
2.3 催化剂加氢性能评价
以萘加氢为模型反应,考察了两种催化剂的催化活性(图10),在0.5Pt/HY催化剂上反应1h后萘的转化率和十氢萘的产率均达到了99.9%,远高于0.5Pt/HY-T 的94.0%和19.2%,60℃下的TOF 值高达2171h-1,显著高于同类催化剂[27-28]。0.5Pt/HY 催化剂上较高的Pt原子利用率以及一定比例的Pt2+/Pt0在吸附活化反应分子中的协同作用是高效催化萘深度加氢的关键因素。可能的加氢机理如图11所示,其中缺电子的Pt2+有利于反应物的吸附,而d 轨道电子充盈的Pt0更利于H2的吸附活化[29]。通常来讲H2的活化是加氢反应中的限速步骤[30],因此加氢过程中需要较高的Pt0占比,虽然0.5Pt/HY-T 上Pt0占比更高,但由于Pt 粒径较大导致大量的Pt0埋藏于颗粒内部无法参与反应,严重降低了原子利用率。且已有文献报道,Pt纳米粒子与氢分子的结合力较弱,不利于它们的活化[31]。不同于0.5Pt/HY-T,0.5Pt/HY中Pt团簇分布均匀且粒度适中更利于氢的吸附活化。综上,0.5Pt/HY催化剂在Pt价态分配及粒度分布上的显著优势为萘、氢分子提供了适宜的吸附活化路线,使得萘的深度加氢得以高效进行。此外,0.5Pt/HY 上顺反式十氢萘的比为5.7,与0.5Pt/HY-T(5.6)基本一致,说明Pt 粒径大小不会对其比例产生影响。
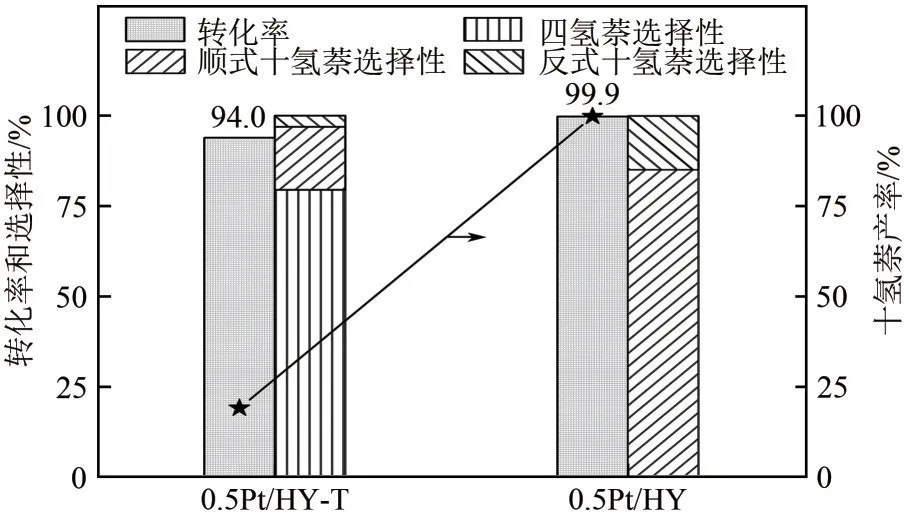
图10 0.5Pt/HY和0.5Pt/HY-T催化剂上的萘加氢反应结果
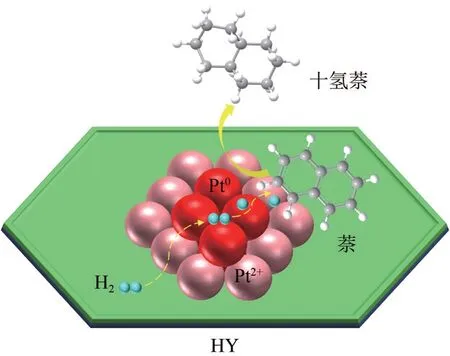
图11 萘和氢分子在0.5Pt/HY催化剂上的吸附活化机理
进一步考察了温度对0.5Pt/HY催化活性的影响(图12),随着反应温度(40~100℃)的升高,萘的转化率(14.2%~99.9%)逐渐升高,同时十氢萘的产率也从1.2%升高至99.9%。整个过程中仅当萘的转化率接近100%时,十氢萘的选择性才开始显著上升。萘加氢反应是可逆连串反应,在单环芳烃上加氢比较容易,从热力学上来看,萘结构比较稳定,只有当其中一个环完全加氢饱和后,余下的环才能开始加氢,受到反应可逆性和芳环稳定性的影响,萘一步加氢生成四氢萘比四氢萘加氢生成十氢萘容易[32-33],所以在反应温度低于60℃时四氢萘的产率远高于十氢萘。从动力学上来看,适度提高反应温度可加快反应速率,利于萘的深度加氢,因此当反应温度高于80℃时十氢萘的产率远高于四氢萘。
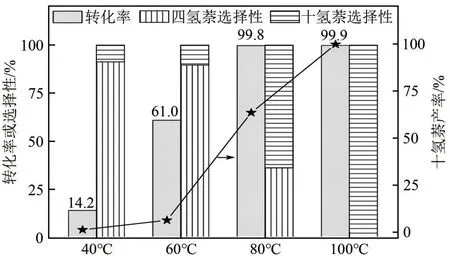
图12 不同温度下0.5Pt/HY催化剂上的萘加氢反应结果
对反应后的0.5Pt/HY催化剂进行了XPS及CODRIFT 表征以考察其稳定性,如图13(a)所示,反应后的0.5Pt/HY 中Pt2+所占比例为25.5%,略低于反应前(33.9%),反应中氢气作为还原剂使得少量Pt2+物种得到电子后转化为Pt0物种。结合图13(b)可知反应后的0.5Pt/HY中2162cm-1处的单原子峰消失,2098cm-1处的单原子峰增强,虽然部分Pt2+对应的单原子峰发生了红移但并未发现Pt 团簇峰的增长,表明反应过程中仅有少量的Pt 物种在价态上发生了变化但聚集状态却基本没有改变。可见催化剂上Pt 物种作为加氢反应的活性中心具备较好的稳定性。为进一步评估催化剂的重复利用性,考察了催化剂在八次萘加氢反应中的催化活性(图14),结果显示,前6 次重复中萘转化率及十氢萘产率均为100%,之后十氢萘产率小幅下降(第7次为99.8%、第8次为99.4%),证明该催化剂具备优异的重复使用性。
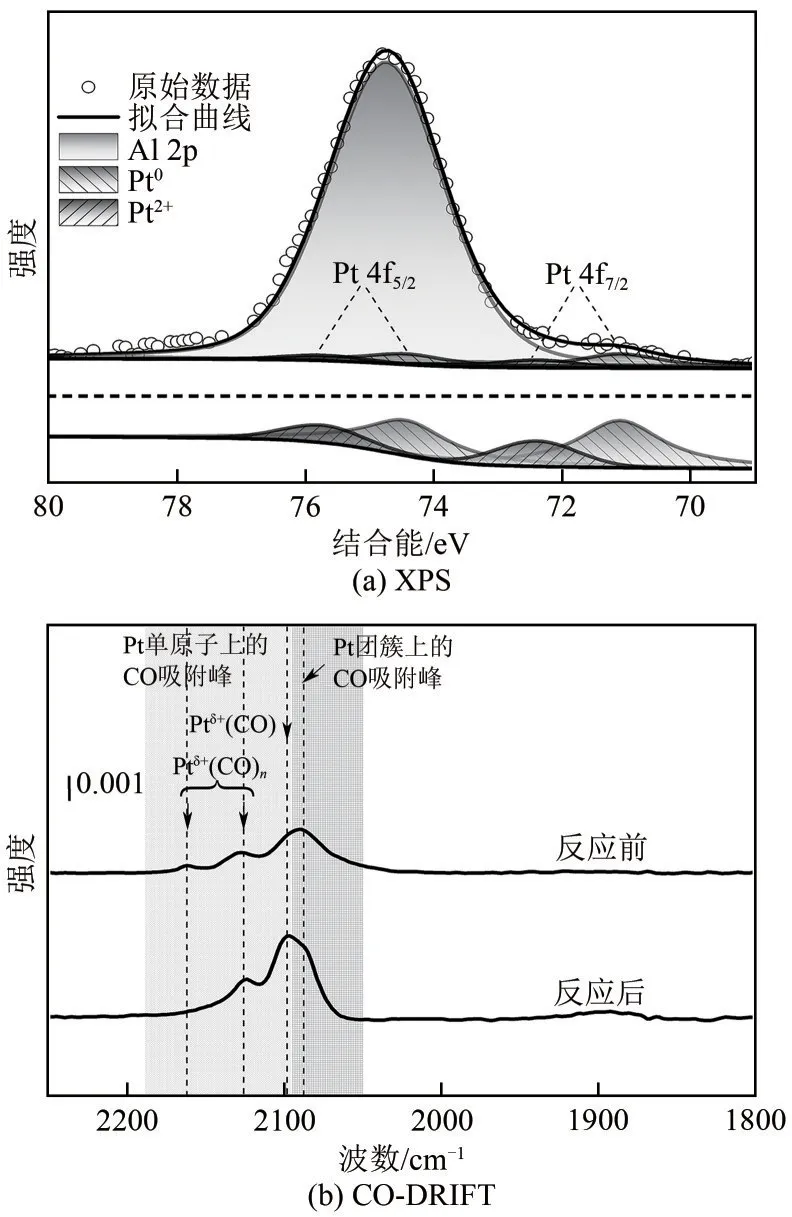
图13 反应后0.5Pt/HY催化剂的Pt 4f XPS光谱图和原位CO-DRIFT光谱图
3 结论
论文以乙酰丙酮铂作为前体负载到HY沸石孔道中,通过高温快速热处理分解有机配体释放出Pt物种,制备了高度分散的负载型Pt 基催化剂。和传统的持续煅烧不同,在配体去除的过程中,通过快速升温可以将与铂物种配位的有机配体快速脱除,有效避免了Pt 物种在长时间的高温环境中发生迁移和聚集,从而有助于获得高分散的Pt物种。0.5Pt/HY催化剂上Pt物种高度分散、粒径较小且部分显示正价态,在萘深度加氢的反应中表现出了优异的催化性能。在温和的反应条件(H23MPa、100℃、1h)下,0.5Pt/HY 对十氢萘的产率达到99.9%,远高于0.5Pt/HY-T催化剂(19.2%)。0.5Pt/HY催化剂具有较高的催化稳定性,经八次循环使用,其活性和选择性均未发生明显变化。催化剂中部分Pt2+物种有利于萘的吸附,而Pt0物种d 轨道电子充盈利于H2的吸附活化,二者的协同作用提高了萘深度加氢的催化效率。
猜你喜欢
天津医科大学学报(2021年1期)2021-12-05
湖北农机化(2020年4期)2020-07-24
工程与建设(2019年5期)2020-01-19
材料科学与工程学报(2016年4期)2017-01-15
现代检验医学杂志(2016年5期)2016-08-20
合成化学(2015年4期)2016-01-17
无机化学学报(2014年6期)2014-02-28
无机化学学报(2014年5期)2014-02-28
华东理工大学学报(自然科学版)(2014年5期)2014-02-27
茶叶通讯(2014年2期)2014-02-27
