
人工智能驱动化学品创新设计的实践与展望
2023-10-07 12:34吴正浩周天航蓝兴英徐春明
化工进展 2023年8期
吴正浩,周天航,蓝兴英,徐春明
(1 美国西北大学土木与环境工程系,伊利诺伊州 埃文斯顿 60208-3109;2 西交利物浦大学化学系,江苏 苏州 215123;3 中国石油大学(北京)碳中和未来技术学院,北京 102249;4 中国石油大学(北京)重质油国家重点实验室,北京 102249)
当今,化学品广泛应用于医药、能源、材料、食品等领域,新型高性能化学品的研究和开发引起了密切关注。但化学品的设计和制备是一个复杂过程,涉及大量高维空间的研究参数。传统的化学品设计过程通常是通过将化学原理与工程技术相结合,基于理论与直觉来调整配方和参数以指导实验设计,从而寻找最优方法来设计和开发高性能、高效率和环境友好的新型化学品。但这些传统的“试错”方法普遍受到“维度灾难”的困扰,不仅需要付出极高的时间和材料成本,也难以保证在几乎无穷的材料设计参数中寻找到最优解,更难以阐明微观结构与关键性能和机制之间的关联,极大地限制了新产品的有效开发和化学工业的科技进步。相比于实验技术,多尺度计算机模拟技术为充分探索化学品的结构-性能关系和分子机制提供了一个高效路径,并具有更多的“接口”与人工智能(AI)进行结合:一方面,AI 提供了将多尺度模拟结果中复杂参数空间映射到目标函数的非线性方法,保证了在化学品复杂材料结构空间中进行高效的性能(构效关系)映射的可能性;另一方面,通过将遗传算法、主动学习和深度生成模型等机器学习算法融入化工材料设计过程,提供了根据化学品目标性能获得相应结构参数的高效反向设计方法。
因此,将AI有效融入到多尺度模拟计算中,有望克服化学品创新设计中的瓶颈问题,是“Al for Science”这一理念的典型代表。然而,这一融合过程涉及的研究方向和内容繁多。本文作者团队以化学品创新设计的可行性、精准和高效为目标,认为除了大家所熟知的构建精准机器学习构效预测模型,以下三个方面的AI 驱动研究应该摆在更为重要的位置(图1):准确快速的多尺度模型;高效的材料正向、反向设计方法;基于AI特性的科学计算软件。本文将首先从计算机模拟方法的创新出发,围绕化学品中典型的高分子材料,例如高分子相容剂、高分子导热材料进行讨论。同时关注AI驱动化学品创新设计,强调在AI时代中,创新计算机模拟基础设施对未来化工研究与发展的重要性。

图1 人工智能驱动化学品创新设计的三个重要方面
1 AI辅助的多尺度模型与分析
现实中,化学品的设计需要借助表征手段分析原子和分子的相互作用关系,以阐明相关机制。但在多尺度计算模拟中,研究者首先要基于符合物理规律的相互作用关系构建准确的原子和分子模型,以进行化学品的创新设计。在化学工程系统与化学品设计中,通常涉及从材料组分的分子到反应器的工业装置等多层次时空尺度,通过多尺度计算机模拟技术(例如,从量子化学到分子动力学再到连续介质力学),可以实现自下而上,以电子、原子等微观粒子为基础,重新认识和重构传统化学化工过程。然而,经典的计算机模拟,虽然在部分领域显示出卓越的作用,如单分子功能预测,但仍然无法有效地应用于与实际相近的真实系统,因为其所需要的计算资源随模拟体系的增大而指数级增加。近年来,人工智能(AI)技术在自然科学、计算科学等学科中的融合发展为改进并加强多尺度模型构建和采样速率,特别是高模拟精度及速度提供了可能(图2)。其中,通过AI技术来帮助发展更全面准确的理论技术来确定粒子之间在各种条件下的相互作用力是改善多尺度模拟计算技术重要的方向之一,以此为新型化学品设计提供更坚实的基础。

图2 人工智能辅助的多尺度模型建立
1.1 全原子机器学习势能
准确表示微观粒子(原子)间复杂的相互作用(势能面)对于使用分子动力学模拟预测材料性能和设计至关重要。虽然,在Born-Opperheimer约化条件约束下,可以通过量子力学计算电子云之间的相互作用力得出原子间作用力,即从头算分子动力学(ab-initio MD)模拟。但是,ab-initio MD 所需的计算资源极为昂贵(关于粒子数指数级增长)。因此模拟的体系往往局限于10~100 原子,限制了其在化工材料领域的广泛应用。最近,机器学习的快速发展提供了高效准确拟合高维势能面的可能性,能够用较低的计算成本提供实现较高的准确性。例如,Deep Potential 团队成功地以ab-initio 的精度在分子动力学中模拟了百亿原子级别的体系,展现出了机器学习势能(machine-learning force-field,MLFF)的优势与潜在的应用前景。
然而,MLFF 在化学化工研究中的普及仍存在挑战。目前常规的机器学习架构主要适用于文字、图片、视频等一维和二维的对象,如何精确地表达三维空间中的原子,并使其符合物理定律,仍是机器学习领域的一个难题。另外,使用现有的高精度的ab-initio MD 技术来标记数据较为耗时,如何使用更少的数据训练以获得性能较好的MLFF是一个亟需解决的问题。为了更好地评估MLFF,找出其中对性能至关重要的特征,从而推进MLFF领域更好的发展,本文作者团队最近参与的一项工作[1]在材料、蛋白质、小分子等多种体系上测试评估了一系列具有不同特点的MLFF,包括DeepPot、Dimnet、Schnet、Nequip等。这项工作提出了三项新的评估MLFF 的标准:①由模拟轨迹计算的观测量;②模拟的稳定性;③ MLFF 的计算效率。与现行MLFF评估和验证标准要求训练模型的能量/力误差尽量小不同,这项工作明确指出,MLFF 训练时的能量和力误差的数值大小并不一定与其在模拟中的实际表现有明显关联。因此,需要重新考虑MLFF模型的评估标准并在评估模型时综合考虑实际模拟的观测量、稳定性和计算速度,以确保MLFF模型的可靠性和有效性。在评估的一系列MLFF中,具有O(3)对称性的Nequip,即在神经网络操作中内生地嵌入了三维空间中分子的平移、旋转、翻转等对称性,在各项指标和体系中的综合表现出色。经过细致系统的分析并结合最近一项有关机器学习表达能力的研究,机器学习架构在三维空间中分子的多体效应(many-body effect)等的表达能力,特别是对于物理对称性群(symmetry group)的考虑,是MLFF在真实模拟条件下性能的关键。基于这些理解,本文作者团队相信深入研究发展MLFF,将有助于进一步提高其在实际化学化工过程中的应用价值。
1.2 机器学习粗粒化模型
尽管相对于ab-initio 模拟方法,MLFF 全原子模型的计算速度已经提高了数倍,但是其仍受限于较小的系统尺寸和较短的模拟时长(约10nm和1ns)。为了进一步提高模拟的速度,一个广泛使用的概念是粗粒化(coarse-graining,CG)。粗粒化CG 是多尺度模拟中最重要的概念之一,CG 模型也是化学化工领域模拟的重要的组成部分[2-3]。在CG模型中,几个原子或者分子被划进一个“超级”粒子,以减少所需要模拟的粒子数,从而可以在更大的时空尺度上进行分子动力学模拟,使理解和预测与实际化学化工系统更相近的尺度上的机理与过程成为可能。
然而,训练准确的机器学习CG 模型通常需要比机器学习全原子模型更多的数据。这是由于在将聚合原子聚合为粗粒化粒子的过程中引入了额外的误差,增大了学习的难度。因此,获取大量高质量数据是开发可靠高效的机器学习粗粒化模型的关键之一。与此同时,自上而下地基于实验数据建立准确的粗粒化模型亦是一项极有意义且颇具挑战的工作。自动微分作为机器学习核心组成部分,最近被引入分子动力学中,以高效准确地开发势能模型。在最近的一项工作中,本文作者团队使用自动微分技术将整个分子动力学模拟可微分化从而使得从模拟轨迹直接对势能函数计算梯度成为可能[4]。将可微分模拟与随机梯度下降优化方法相结合,本文作者团队开发出一种新的方法来优化势能函数,展示了如何使用可微分分子动力学模拟方法,从高质量的实验数据中自上而下地学习出液态水分子温度可转移的粗粒化模型。这为未来构建更可靠的通用粗粒化模型奠定了坚实的基础。因此,将机器学习方法与以粗粒化模型为代表的多尺度模拟策略有机地结合,通过引入高质量的实验和模拟数据,可以促进化学化学品快速高效的多尺度设计与开发。
1.3 机器学习辅助模拟分析
机器学习方法不仅可以用于构建物理模型,还广泛应用于进行模拟结果的分析。在材料研究中,构建结构与性质之间的精确关系(即构效关系)非常关键。机器学习因其强大的适应能力而成为构建构效关系的强有力技术。在最近的一项研究中,本文作者团队成功运用高效的耗散粒子动力学模拟技术,预测了数百种不同接枝情况下共聚物刷的相容性。针对这些高度复杂的数据,本文作者团队运用机器学习技术有效构建了结构和性质之间的关系,为未来高性能聚合物相容剂的开发提供了高效直接的预测模型。另外,分子动力学模拟的结果需要从非常复杂的轨迹、能量等数据中进行分析和发掘。机器学习因其强大的降维、拟合等能力,已经成为了分析分子动力学模拟数据的强有力工具。例如,本文作者团队利用神经网络以脂质分子原子轨迹作为输入,准确预测在不同酒精浓度下脂质分子的构象性质[5],从而帮助判断冠状病毒模型细胞膜在酒精作用下的稳定性。神经网络高效准确的预测能力,使得快速分析数成百上千个组成成分的细胞膜成为可能,为开发高效的病毒酒精消毒液提供了理论基础。随着机器学习在分子动力学模拟数据分析中的广泛应用,分子模拟在实际场景中的应用将得以更快速地推进。
2 材料设计方法
众多先进化工技术的发展是以特殊性能材料为核心。例如,膜分离技术作为支撑水资源、能源、环境、传统产业改造等领域的战略性高新技术,亟需新型具有高选择透过性的高性能聚合物材料设计与制备进行支撑。然而,聚合物材料的结构设计和性能优化是一个跨越多个尺度的复杂系统,看似微小的结构变化,如在高分子链中增加一个支链或单体序列的差异,都会对性能产生巨大影响。为此,有针对性地开发高效材料设计方法成为关键所在。为了解决这一挑战,本文作者团队基于正向设计和反求设计两种研究思路,针对聚合物材料这一典型化学品,探索构建智能研究框架,进行AI 驱动的设计和结构调控,见图3。

图3 人工智能辅助的材料设计方法
2.1 正向设计
随着计算能力和基于物理模型的迅速发展,计算机模拟,特别是分子模拟技术,可以高效测试化学化工材料的性质并进行预测。总的来说,与实验相比,计算机模拟作为一种正向设计的工具,可以极大地提高材料研发和设计的效率,同时能够降低成本。
以聚合物为例,将已有聚合物进行共混可提高聚合物材料力学性能、加工性能,降低成本,并扩大使用范围,是实现聚合物改性和生产多功能新型聚合物材料的重要途径之一。然而,绝大多数聚合物是不相容的,导致混合产品的机械性能差、界面张力高。研究表明,在共混物中加入与其具有相同化学组分的共聚物是实现突破原有体系热力学极限、开发高性能聚合物的有效方法。但对于共聚物来说,微小的结构变化(如嵌段连接方式或序列的差异)都会对性能产生影响。因此,亟需开发一种能够从繁多结构信息中解析与辨认其关键作用的相容性描述符,以便实现高效、合理地设计高性能相容剂的分子结构。
本文作者团队首先通过耗散粒子动力学模拟(DPD)研究了线型嵌段共聚物在不相容聚合物相之间的相容性能[6],并以此解析了相容剂结构特性,建立了幂律拟合模型,实现了线型嵌段共聚物的相容效率随聚合物化学、分子结构和共聚物分子数量的变化预测。基于此研究,本文作者团队还针对由骨架和多个侧链组成的更复杂拓扑结构的接枝共聚物[7],结合热力学分析和界面模型计算,关联界面张力随接枝共聚物多样性结构参数变化的关系,从而辨识出起主要相容作用的结构描述符,并以此为基础开发了DPD/ML的智能研究框架,实现了在分子水平上准确预测具有多个描述符(如分子结构和化学成分)的特定接枝共聚物的相容性效率,并基于不同描述符的重要性等级,开发了ML模型的机理解释方法。以此为基础,本文作者团队成功实现了高相容性共聚物的分子量、拓扑结构和序列的精准设计,开创了以功能基元-序构为核心的相容剂正向设计的普适方法,创新发展了共聚物拓扑结构解析方法,剖析了界面增容时共聚物的空间分布机制,结合热力学剖析构建机器学习辅助的研究框架,提出了共聚物增容信息的描述符辨认方法,并以此进行高相容性共聚物的分子量、拓扑结构和序列的精准设计,从而开创了一种以功能基元-序构为核心的相容剂正向设计的普适方法。
2.2 反求设计
反求设计,即如何从复杂的材料设计参数空间中找到对应材料目标性质的参数,对实现高性能化学化学品的创新设计十分重要。以下本文作者团队将以高性能聚合物导热性质为实例探讨如何将AI结合理论与模拟进行有效的材料反求设计。
共聚物材料的低本征导热性会导致积热,进而引发材料溶胀、产品性能降低。高效开发高本征导热性共聚物材料已经成为储能技术发展的强烈诉求和重要科学问题。本文作者团队瞄准这一科学问题,提出从聚合物材料的单体排列具有基因特性这一特点出发,利用材料基因组策略进行聚乙烯-聚丙烯(PE-PP)高导热性材料设计。反求设计通过对已知的多种材料中主动搜索未知的最佳材料,相比基于结构-性能关系的正向筛选设计,降低了对数据库本身的需求,具有较好的研究前景。
本文作者团队采用遗传算法和分子动力学模拟的组合框架来设计PE-PP 共聚物,基于反求设计获得具有高热导率的特定序列共聚物:明确不同序列的PE-PP 共聚物的热导率分布规律,首次获得了高出常规二嵌段共聚物40%的非直觉高导热性共聚物(non-intuitive),进一步解析解析发现体密度、链构象和振动状态密度不能准确解释导热性变化,提出了基于分子作用力和振动函数分析方法,量化了阐明单体序列的变化和热能传输效率的构效关系,提供了将MD模拟与遗传算法相结合来设计新型材料的应用实例[8]。本文作者团队创新发展基于材料基因组策略的高导热性聚乙烯(PE)-聚丙烯(PP)材料的理性设计,揭示基元PE-PP序构材料中蕴含的导热变化规律,建立了超越人工筛选可探索范围的变革性聚合物材料反求设计方法。
3 数据管理与软件开发
现代化的开源数据和高效便利的软件开发是将AI 技术运用于实际的关键。针对化学化工领域AI驱动的创新设计,本文作者团队将从数据管理和科学计算软件开发两个方面展开讨论(图4)。
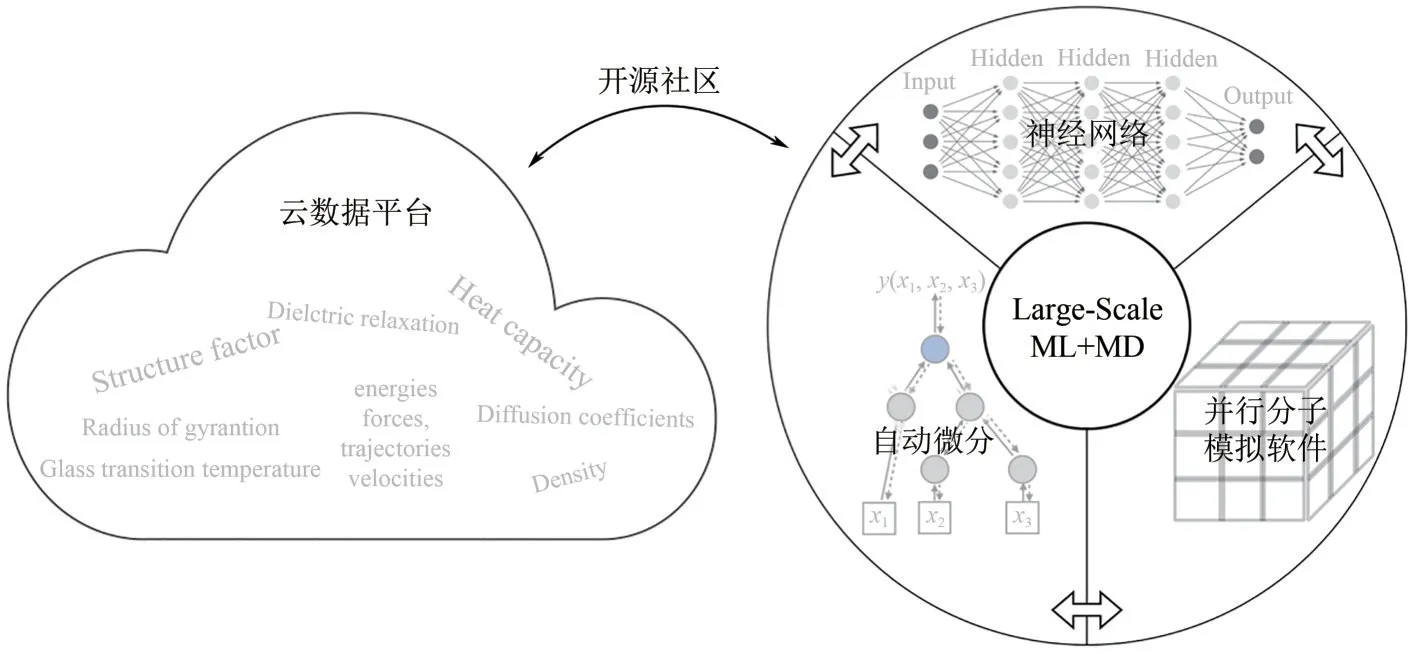
图4 网络基础设施:以数据驱动的材料化学信息云平台和开发与机器学习相适应的科学计算软件
3.1 数据管理的云平台
随着近年来计算和实验研究中高通量工作流程的发展,物理、化学和材料科学领域产生了大量的数据。为了更好地利用这些数据进行分析、验证和进一步发展,材料科学领域的开放数据库已经启动,如材料基因组计划(Materials Genome Initiative)、材料云(Materials Cloud)和聚合物数据库(PolyInfo),这些数据库均存储了来自实验和计算的材料结构及其相应的属性。将机器学习(ML)整合到分子模拟中,促使许多数据驱动的方法开发基于ML的分子模拟模型。这些模型通常是通过输入大量的数据来训练的,如系统能量和原子力,这些数据由昂贵的高保真模拟内部产生。一些开放的数据库已经启动,以加速模型的发展,促进透明度和可重复性。然而,与其他领域相比,由于其固有的复杂性,化学化工领域的高质量数据集仍然有限。因此,建立一个收集和重用超级计算机本地存储的模拟数据的平台,不仅可以加速例如聚合物材料分子模拟的发展,而且可以节省资源,实现可持续性。结合上文提及的数据驱动多尺度模拟与分析技术和材料设计算法,如何开发一个高质量的云平台,以改善开发化学化学品的数据存储和共享将会是下一步工作目标。为了促进数据驱动的化学化工材料设计,本文作者团队计划创建一套数据存储与分发的算法,包括标准数据格式和开放代码库,以处理来自不同尺度的实验和模拟方法的数据。一个统一的数据存储和标准化流程可以促进模拟和实验的融合,从而在基础设施上加快数据驱动地化学化工材料的创新设计。
3.2 科学计算软件
科学计算软件在现代科学的不同领域,从化学和生物学到物理学和材料科学,一直是一个宝贵的工具。以分子模拟为例,在过去的30 年里,人们为开发高效的计算算法和高质量的分子模拟包(如LAMMPS 和HOOMD-BLUE)付出了巨大努力。基于先进高效语言进行的并行算法的分子模拟软件的开发使得多达数十亿的原子分布在数十万个计算节点上的计算成为可能,拉近了与实际应用场景的时空尺度。在此方向上,本文作者团队基于现代的、面向科学计算的高性能动态高级程序设计语言Julia开发了应用于软物质的大规模并行混合粒子场分子模拟软件RobertoMD.jl[9]。然而,这些经典模拟器通常有一个巨大的专业代码库,如物理方程的手工梯度和用于GPU加速的定制CUDA或OpenCL内核,用C++或Fortran编写,使得它难以适应快速发展的算法和硬件的进步。机器学习正在成为科学计算的一个重要方向,可以使分子模拟更加准确和高效。其中大部分的成功都归功于自动微分的应用,它通过按照链式法则使其可以准确计算任意计算机程序的梯度。基于机器学习框架的端到端(end-to-end)可微分(differentiable)分子模拟器,如TorchMD和JaxMD,最近已经被引入化学、物理、生物等的科学界。虽然目前更多的是作为一个原型设计平台,但这些可微分的科学计算程序已经在各个领域展示了它们的优势,如分子模拟其中的机器学习势能函数的开发和部署。与LAMMPS 这样的传统分子模拟软件相比,可微分的分子模拟程序可以很好适应机器学习势能函数,而不需要费时且易出错地手工推导,如神经网络的梯度,或者与外部具有自动微分功能的代码库通信。开发一个完全端到端可微分的分子模拟器,并支持高度可并行处理的多个计算单元,将对无缝整合机器学习技术到大规模分子模拟中起到关键作用,缩小生产级分子模拟和机器学习技术之间的差距。
4 结语与展望
AI 与各个学科的融合发展已经成为一个热门话题。从学术研究角度来看,几乎所有的自然学科都有科研人员尝试将AI 这个工具引入到其研究领域,以试图探索新的研究方向[10]。但是,这一过程当前也存在着较多问题。目前很多研究者并不关心AI 算法在其自身领域的适用性和完善性,而是简单地将AI 套用进原有科学问题中,利用AI进行简单的数据处理,例如将原来用线性回归解决的问题改成多层神经网络预测,对进一步理解科学问题背后的微观原理作用有限。在这些AI 应用场景之外,本文作者团队认为,在化学品的创新设计中,AI的优势体现在“生成”:即通过结合多尺度模拟,实现在已有研究结果中“生成”新特性和新产品,本文所讨论的多尺度模型构建、高效设计方法和软件开发将加速这一过程的实现。另外,将前人研究已经确定下来的物理作用和规则加入到AI 算法设计过程中,增加AI 模型的可解释性与转移性,也是在未来深入融合AI 与特定的科研领域的关键。综上,本文主要介绍AI 方法在化学品设计与开发中的应用实践,认为AI 驱动的科学研究应该是将AI 和科学研究进行高度关联和耦合,旨在抛砖引玉,共同推进高性能化学品的创新设计发展。
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29
内蒙古民族大学学报(社会科学版)(2020年2期)2020-11-06
劳动保护(2019年3期)2019-05-16
太空探索(2016年5期)2016-07-12
安全(2015年6期)2016-01-19
化工进展(2015年3期)2015-11-11
中国科技信息(2015年17期)2015-11-02
中国塑料(2015年1期)2015-10-14
材料研究与应用(2015年4期)2015-08-23
时代英语·高三(2014年5期)2014-08-26
