
融合基因分析方法在肿瘤研究中的应用与发展
2023-09-20 10:00张军炜管欣超杨巧媛
广州医药 2023年8期
张军炜 管欣超 刘 韬 杨巧媛
广州医科大学公共卫生学院(广州 511436)
现代研究普遍认为,肿瘤的发生与遗传物质的改变密切相关。与肿瘤生长相关的基因主要有原癌基因与抑癌基因,分别参与细胞生长发育与抑制生长的调控过程。融合基因由两个或以上基因的序列发生断裂并结合产生,大多数融合基因的产生都与染色体的结构发生畸变有关,而染色体异位在肿瘤发生之初发挥着重要的作用[1],因染色体发生重排、缺失、异位和颠倒而产生的融合基因驱动了许多肿瘤的发展,这也被认为是肿瘤发生的重要因素。融合基因最早于白血病样本中被首次发现[2],这是由9号染色体上的ABL基因与22号染色体上的BCR基因相互融合形成,并形成费城染色体[3]。
已有的研究中有越来越多的疾病被发现与融合基因有关,如肺癌[4]、乳腺癌[5]、甲状腺癌[6]、前列腺癌[7]、白血病[8]等,因此对于融合基因的检测及预测对临床上疾病诊断及治疗具有重要意义。由于融合基因是由两个或以上基因的编码区首尾结合形成的嵌合基因,其往往能够转录出相应的嵌合RNA,但嵌合RNA并不仅仅通过融合基因转录生成,反式剪接也是嵌合RNA产生的主要机制之一。因此通过对转录组进行测序的方法识别出嵌合RNA后还需要再进一步对基因组进行分析,以此确定该RNA为融合基因的产物。临床上对于融合基因的识别及检测主要包括:荧光原位杂交(fluorescence in situ hybridization,FISH)[9]、逆转录聚合酶链式反应(reverse tran scription polymerase chain reaction,RT-PCR)[10]以及测序技术如sanger测序[11]和高通量测序技术(next-generation sequencing,NGS)[12]等方法,而关于融合基因的预测则更多需要借助其他生信工具进行,大多数对于融合基因的预测方法都需要预先准备好读长不一的序列。
1 融合基因相关产物
1.1 嵌合RNA与融合蛋白
目前的研究发现,几乎所有的融合基因均能够产生相应的线性转录本(即嵌合RNA)。嵌合RNA由两个或两个以上基因的外显子通过某种机制发生剪切组合而成,研究认为目前嵌合RNA的形成机制主要包括顺式剪接,反式剪接以及融合基因的转录。传统意义上,顺式剪接是来自同一个基因的不同外显子彼此连接的过程,当两个或多个同一来源的前体mRNA剪接在一起即可形成嵌合RNA。反式剪接是两个或多个不同来源的前体mRNA被剪接在一起的过程,该过程同样能够产生嵌合RNA。顺式剪接和反式剪接均发生于转录后RNA剪接的过程中,而由于染色体发生重排等原因导致基因发生融合进而产生嵌合RNA的过程则发生于转录以前,这类机制所产生的嵌合RNA往往具有较高的表达丰度[13]。与普通基因转录得到的mRNA类似,部分的嵌合RNA也可翻译出相应的蛋白质(即融合蛋白),而融合蛋白在临床上具有较高的治疗及诊断意义。正如人类发现的第一个融合基因BCR-ABL,它能够导致人类发生慢性粒细胞白血病[14]。随后科学家们又陆续发现了许多由融合基因产生的融合蛋白如RET-CCDC6[15]、EML4-ALK[16]等,这些蛋白在肿瘤的发生和发展过程中发挥了重要作用,同时也为肿瘤检测与靶向治疗提供了方向。
1.2 融合环状RNA
与正常基因类似,融合基因在转录的过程中也能通过目前尚未完全阐明的机制产生融合环状RNA。环状RNA是一类产生于转录之后,不具有5'末端帽子和3' 末端poly(A)尾巴、并以共价键形成环形结构的非编码RNA分子,一般不易被RNA外切酶或核糖核酸酶R降解,因此稳定型比线性RNA高。环状RNA广泛存在于人体细胞当中,由特殊的可变剪切产生,常常富集于外泌体中并且能够从体液中提取出来,结合其具有肿瘤特异性的特点,现常被用作潜在的肿瘤生物标志物[17]。在人体细胞中,环状RNA可以通过与相应的RNA结合蛋白相互结合,从而影响相关基因的表达,部分环状RNA也能够通过与蛋白的相互作用来抑制翻译的进程。融合环状RNA的产生与普通环状RNA的产生机制类似,其同样为一种由转录后的剪接事件产生的内源性非编码RNA,也与环状RNA一般具有一样的特性和功能,这提示融合环状RNA的存在可能与癌症的发生具有密切的关系,许多研究也正在陆续揭示二者的关联,越来越多以融合基因为主要驱动因素的癌症研究中均发现了相关的融合环状RNA,如WU K等人[18]发现在非小细胞肺癌中,SLC34A2-ROS1融合基因可以产生名为F-circSR1和F-circSR2的融合环状RNA,以及在GUARNERIO J等人[19]的研究中,发现在急性髓系白血病中,PML-RARA融合基因可以产生一个名为f-circPR的融合环状RNA。作为一种较新颖的RNA分子,关于融合环状RNA的研究目前尚有待加强。
2 嵌合RNA分析工具
测序技术发展的同时也促进了基因组学的发展,下一代测序技术已经能有效识别DNA的结构变化以及对不同的转录组进行分析,而其中RNA测序技术可用于在真核生物中识别不同的嵌合转录本。以相应的人类基因组序列作为参考序列,与待分析的RNA read进行比对之后筛选出未能匹配的read,这些read即可认为是嵌合RNA。但并非所有的嵌合RNA均可编码蛋白,研究表明大部分的嵌合RNA均作为非编码RNA而存在[20]。在利用RNA测序技术对融合基因进行检测分析时,这些非编码RNA极有可能影响检测的结果。
对此,Christopher A等[21]利用了高通量测序法,综合了长read和短read的分析,将长read与参考基因组进行比对之后,筛选出仅有部分读长与参考基因组匹配的read作为候选嵌合体;再利用Illumina等平台从相应的序列数据中生成短read,与参考基因组比对后筛选出包含有两个基因片段的read,将这些read与候选嵌合体综合分析,得出部分序列相同的read,其能够包含同样的融合基因断裂点且属于候选嵌合体之一,降低了无效嵌合体的影响及融合基因分析过程中的假阳性事件的发生。现阶段许多针对融合基因的研究工具,基本都围绕RNA序列展开,近年来也诞生了许多围绕机器学习以及神经网络等方法而开发的工具。
2.1 传统RNA-seq分析工具
融合基因的断点多发生于内含子区域,故对于融合基因的分析多从RNA的层面出发,目前常用的相关算法工具主要有SOAPFuse,InFusion,STAR-Fusion和JAFFA等工具,大多数的工具遵从如图1的基本流程,这些工具都具有速度快、准确性良好等优点,同时又具有各自的特点。SOAPfuse和InFusion均能从RNA序列中高效识别出嵌合转录本,前者作为较早期开发的工具,现多与其他工具一同运用或用于与其他分析工具进行比较[22-25],后者能够从RNA序列中检测出基因间非编码区域的融合事件。若在输入的read中存在包含融合剪切位点的序列(SPLIT read),或是由双端测序产生的,含有未测序的序列且跨越了剪切位点的序列(BRIDGE reads),InFusion能够利用其新的算法对这些read进行聚类并重建融合转录本,并在结果中报告相应的基因区域和断点的位置,以及相应融合位点的序列[26]。
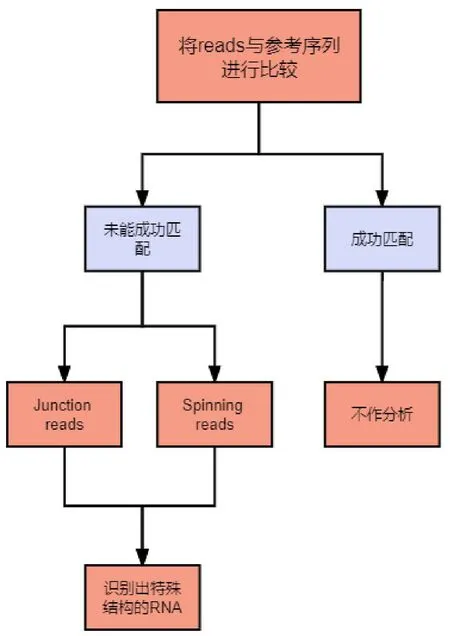
图1 嵌合RNA分析工具的基本流程
STAR-Fusion是建立在STAR基础之上的工具,它能够应用于融合转录本的预测,是目前主流的RNA序列比对软件之一。STAR具有运行速度快、准确的优点,而STAR-Fusion则继承了这些优点,同时在模拟数据和从细胞系中获取的序列数据中仍具有较好的灵敏度和精确度[27]。在一项多种工具比较的研究中,STAR-Fusion具有较高的排名,在大多数情况下能够保证较高的预测精度[28],但STAR-Fusion的运行需要消耗较高的内存,对硬件有一定的要求。JAFFA同为RNA比对软件,但不同的是其将待分析的转录组RNA序列与转录组作为参考进行比较,且能够应用于不同长度的read,同时能够输出候选的融合事件及其相关信息。在分析的过程中,长度小于100 bp的read能够被JAFFA重新组装为100 bp甚至更长的read,对于超长的read 具有良好的特异性,是利用RNA序列数据对融合基因进行分析工作的高效工具,在许多融合基因的研究中被广泛运用[29-30]。

表1 不同嵌合RNA分析工具的比较
2.2 其他分析工具
除此以外,根据不同用途目前也开发了一些不同的算法工具,如为了高通量药物筛选而设计的一套检测算法Arriba[31],它能够在短时间内根据输入的RNA序列进行演算识别,随后输出的结果具有较高的准确性,同时具有计算效率高、高敏感性的优点,能够降低因样品纯度较低带来的影响。在与deFuse、TopHat的比较中,Arriba的可靠程度最高,在15个融合事件的测试中灵敏度达到了80%[32]。此外,Paul Kerbs[33]等人也利用Arriba和FusionCatcher的辅助验证对RNA测序技术和其余标准的诊断技术进行了比较和评估,取得了较好的预期效果。Fcirc则是另一款能够识别出因融合产生的线性或环状RNA的工具,其基于Python开发,能够快速分析输入的RNA序列。与STAR-SEQR和Arriba相比,Fcirc具有更高的精度,同时还有召回率(Recall)良好、分析时间短的特点[34]。在蛋白组学领域,有学者开发了FusionPro,这是一类蛋白质组学工具,它能将蛋白质组和转录组的数据结合分析,并对由融合基因翻译而来的融合蛋白肽段序列进行鉴定,能够用于对融合基因与融合蛋白的研究。Kim[35]等人利用FusionPro在白血病的3个细胞系中分别成功鉴定了82、281以及95个基因融合现象,同时初步揭示了可能存在的基因融合的剪切规律,为融合蛋白与肿瘤发生的关系研究提供了新的方法。
科技的进步促进了人工智能的发展,近年来也逐渐出现基于人工智能所开发的算法工具,如EasyFuse,FusionAI和ChimerDriver等。EasyFuse是基于机器学习开发的一个针对来自于临床样本获取的转录组RNA序列进行预测的方法。在对read进行比对过滤后,EasyFuse仅保留不一致的一对read和比对失败的read,这将能够过滤掉至少90%的read,大大提高了运行速度。该算法具有计算性能高、灵敏度及精度高的优点,能够对多种类型的样本进行预测,David Weber等[36]利用EasyFuse成功表明了融合基因能够提供丰富的肿瘤相关抗原,促进了免疫治疗的发展。ChimerDriver和FusionAI[37-38]都是基于深度学习开发的对融合基因进行预测的算法。前者基于多层感知器的人工神经网络模型,将转录因子和miRNA纳入对融合基因的评估,并以此改进了对基因融合的致癌潜力的预测,同时能够根据相应的转录因子和miRNA的特点将融合基因分为致癌与非致癌[39]。由于基因转录后其调控过程能够影响其致癌的潜力[40],且部分检测到的融合基因,由于其本身并不参与转录与翻译表达的过程,在临床研究(尤其是靶向治疗的研究)中不具备实际意义,因此ChimerDriver能够很好地帮助筛选研究所需的目的融合基因。后者能够利用DNA序列预测出融合基因断点,同时预测该断点是否会成为潜在的融合基因断点[37-38]。由于嵌合转录本的形成原因较多,且融合基因的断点大多位于内含子上,导致融合转录本的断点往往处在两个外显子的边界,因此以RNA序列作为输入数据较难识别融合基因断点的存在。以往的预测算法大多采用RNA序列,因此可能无法获取与DNA双链断裂相关的序列特征,而FusionAI从DNA序列出发,分别以FusionGDB数据库中基于TCGA(The Cancer Genome Atlas)且融合断点位于外显子交界处的序列和同等数量的伪融合断点序列为阳性数据和阴性数据来对模型进行训练,并建立最终FusionAI模型[37]。
3 融合基因的验证及鉴定
得益于测序技术的高速发展,人们发现了大量的嵌合RNA数据,而基于转录组RNA序列展开的一系列对于融合基因的预测技术,得出的结果大多为嵌合RNA。由于嵌合RNA的产生机制有多种,包括基因的顺式剪切、反式剪切以及来自于融合基因的转录等,对于部分方法预测得出的嵌合RNA,暂无法认为其来源于融合基因。因此在实际应用中,还需要进行进一步的实验分析方法来验证嵌合RNA的来源是否与融合基因相关。
3.1 实时荧光定量RT-PCR(qRT-PCR)
qRT-PCR在普通PCR的基础之上增加了逆转录的过程和荧光探针。荧光探针与产物结合后能够产生荧光信号,其强度与扩增的产物成比例增加。目前常用的探针为Taqman荧光探针,其5' 端和3' 端分别带有荧光基团和荧光淬灭基团,当扩增时两个基团互相分离,从而发出荧光信号并被检测系统捕获。在融合基因的检测中,Taqman探针具有特异性高、耗时短的优点,Xiaodong Lyu等[41]利用qRT-PCR对多例急性骨髓性白血病进行分析后显示,qRT-PCR具有良好的诊断效果,与细胞遗传学诊断结果具有高度的一致性,是融合基因检测中的高效方法。但在实际应用的过程中,往往需要研究者预先知晓发生的融合事件,以此来设计相应的引物。
3.2 FISH
FISH技术利用带有荧光基团的单链DNA与目的基因进行杂交,从而对目的基因进行检测。在基因突变、染色体变异等基因组结构研究中拥有广泛的应用,而在融合基因的应用中有实验周期短、特异性强、具有较高的灵敏度和精确度的特点。梁小芹等[42]利用FISH与免疫组化(immunohistochemistry,IHC)一同对ROS1融合型肺癌进行检测并比较了二者的一致性,并未发现二者的检测结果有显著差异,因此FISH在临床上是良好的诊断方法以及检测工具。但由于需要人工在观察,存在着操作复杂且结果主观等缺点。
3.3 IHC
IHC的基本原理是抗原抗体的特异性结合。利用抗原与抗体结合后,发生化学显色并被显微镜或其他仪器捕获,从而能够在细胞层面对融合蛋白进行识别及分析。根据标记物的不同IHC也有不同的分类,临床上目前常用的方法包括免疫荧光法、酶标法等。前者利用带有荧光标记的抗体作为探针,当与所需抗原结合后可发出荧光波长并被检测仪器捕获;后者利用带有酶标记的抗体,与相应的抗原结合后再与后加的酶底物相互作用,释放具有颜色的物质,该物质可于显微镜下观察到。该方法具有操作简单、成本低廉、适用范围广等优点,但也存在敏感度低、特异度差、无法区分不同类型的融合基因等缺点。从肿瘤诊断的角度,IHC检测往往作为基因融合的替代标志物,如在NTRK融合基因的应用中,pan-TRK IHC检测较为广泛,但大多数情况下均用于对NTRK融合的筛查工具[43],即使其在NTRK的检测中具有较高的敏感度,目前仅允许作为临床诊断的辅助工具[44],对于某些呈现弱阳性的样本,可能仍需要通过NGS等其他方法去进一步证实NTRK融合事件的发生。
4 讨 论
从费城染色体的发现开始,融合基因逐渐成为肿瘤学领域研究的热门话题,期间也产生了许多关于融合基因的产生和作用机制的研究,越来越多的作用通路也逐渐被揭示,同时也使得人们对融合基因的认识越来越深入。但即便如此,在测序技术尚未发展成熟的早期,人们对融合基因的识别非常困难。近几年随着测序技术的不断发展,人们获得了大量的基因序列数据,利用此技术也发现了许多转录组的数据,其中包括嵌合RNA。由于人类基因组中含有内含子序列,因此从转录组出发,对融合基因进行分析是目前研究的主要趋势。在嵌合RNA分析领域,有许多不同的生信工具,他们各自因不同的优化和原理具备了不同的优缺点。纵使在实际应用当中并不会全部使用这些工具,但研究者仍能够根据自身的需求以及不同配套的硬件设备去选择不同的分析工具,且总体上看基于嵌合RNA的分析工具正在不断往更高的效率、更良好的准确性发展。由于融合基因并不是产生嵌合RNA的唯一机制,从嵌合RNA出发对融合基因进行研究尚存在困难,后续仍需要对其是否来源于融合基因进行实验验证。主流的验证实验如qRT-PCR,荧光原位杂交和免疫组织化学等虽然都具有较高的准确性,但同时也具有成本较高、试剂昂贵等缺点,这也是融合基因分析的部分局限性所在。
作为癌症的驱动基因,融合基因逐渐成为近年来癌症研究的一大热点。利用基因组学对癌症进行分析,能够根据不同类型的患者进行不同的诊断及治疗,大大提高了诊疗的精确性。新的基因组学分析方法的出现,尤其是针对相关融合基因的预测,对肿瘤研究具有重要意义。无论是过去单靠测序技术发现的融合基因,还是如今依靠RNA测序衍生的一系列生物信息学工具,虽然都存在一些不可避免的缺陷,但都能够用于筛选靶基因以用于分子靶向药物的治疗,在临床上拥有广泛的用途。在前沿领域,还存在许多基于不同理论研究而开发的算法[45-48],如何将理论层面的算法与融合基因分析相结合相信是未来融合基因分析发展的方向之一。在科技逐渐发达的时代,对于融合基因分析的方法将越来越成熟。
猜你喜欢
现代实用医学(2022年10期)2022-12-08
新民周刊(2022年27期)2022-08-01
中国民间疗法(2021年9期)2021-07-22
传染病信息(2021年6期)2021-02-12
初中生世界·九年级(2019年6期)2019-08-15
上海铁道增刊(2017年2期)2017-04-18
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
组合机床与自动化加工技术(2014年12期)2014-03-01
无机化学学报(2014年5期)2014-02-28
