
有序-无序诱导AgBiS2 可见-红外光响应调变机制
2023-08-28 08:37滕洪阳杨竞秀
化工技术与开发 2023年8期
滕洪阳,杨竞秀
(吉林建筑大学材料科学与工程学院,吉林 长春 130118)
红外光的检测是现代工业﹑科学﹑能源﹑医疗和国防应用的基础。近些年来,红外探测器技术已逐渐成为红外技术发展的根本,开发高性能﹑低成本的红外探测器技术已经成为红外技术发展的必然,各种新型红外光电材料的出现不断推动着红外技术的发展。
自1917 年美国的Case[1]研制出第一个Tl2S 光导型红外探测器开始,现代红外探测技术已历经了百余年的发展。19 世纪30 年代,PbS 红外光电探测器的出现[2]及其在二战期间的应用,使得人们意识到发展红外探测技术的重要性,在此基础上,PbSe 和PbTe[3]等多种铅盐类红外材料也陆续被发现并得到应用。但这类材料因热膨胀系数较大而难以与硅工艺集成制备大阵列器件,且量子效率偏低。InSb 半导体[4]在低温下仍具有较好的物理化学性能,但其探测波长只能延伸到中波红外区。1959年, Lawson 等人发现并成功合成的HgCdTe 材料[5],对红外探测技术的发展起到了极大的推动作用,也是目前应用最为广泛的红外材料。HgCdTe 材料由具有负禁带的HgTe(-0.3 eV)和正禁带的CdTe(1.5 eV) 混合而成,是一种具有直接带隙的赝二元化合物材料,通过调整Cd 的组分可以调节材料的带隙,从而实现对整个红外波段的探测[6]。但这种材料存在制备工艺复杂﹑所需操作温度较低﹑成本高昂的问题[7],同时材料中所含的Hg﹑Cd 和Te,都是可能对人体产生危害的物质。1977 年,IBM 研究院的Sai-Halasz 和Esaki 等人提出了Ⅱ类超晶格的概念,在红外探测领域引起了极大的研究兴趣[8]。Ⅱ类超晶格材料是一种可以通过对材料物理与晶格参数的设计和控制,改变其能带结构进而优化其光电性质的材料,近些年来得到了迅速发展。根据理论预测,Ⅱ类超晶格可能具有比HgCdTe 更好的性能[9],但由于起步较晚,目前这些更加优异的性能仍未完全实现。目前Ⅱ类超晶格材料还存在量子效率低﹑器件的暗电流高等问题。除了调控组分﹑晶格等能带调控方式外,通过调节和控制组态(离子占据的有序性)来调控半导体的能带,也是一种常见的能带调控方法。Yang 等人[10]通过第一性原理计算,成功预测了Cs2AgBiBr6有序无序结构的转变温度,并明确指出,通过组态调控,其光响应范围可以在紫外-红外区实现连续调控。
AgBiS2是一种稳定﹑环境友好﹑储量丰富的可见光响应的半导体。目前,AgBiS2的纳米晶已成功合成[11]。最近的研究指出,基于AgBiS2的纳米晶体太阳能电池的能量转换效率已达到9.17%[12]。借鉴组态调控方法,通过改变化合物中Ag+和Bi3+的占据序列,就有可能大范围调控AgBiS2的能带结构。在本文中,通过第一性原理计算,基于准无序结构模型,我们进一步研究了这种可能性。计算结果表明,改变Ag 和Bi 占据位置的无序度,确实可以调控AgBiS2的光学带隙,从而实现其光响应范围的大幅度红移,在红外探测领域显示出巨大的应用潜力。
1 计算方法
本文中的第一性原理计算采用了基于密度泛函理论(DFT)框架下的VASP 软件包[13-14]。软件包中内置的投影缀加平面波赝势(PAW 赝势)[15]用于结构优化。此外,结构优化采用基于Perdew-Bueke-Ernzerhof (PBE) 的广义梯度近似GGA[16]作为交换关联泛函,平面波的截断能设置为400eV,优化的几何收敛标准是 0.01eV・Å-1,晶格常数和离子位置全部弛豫。采用8×8×8 Gamma 样式的K 点网格优化完全有序结构的原胞。为使K 点的密度保持一致,构建的完全无序结构进行优化时的K 点,采用了2×4×4 Gamma 样式K 点网格。由于PBE 泛函通常会低估带隙,为了保证计算结果的准确性,进一步采用杂化泛函HSE06[17](Heyd-Scuseria-Ernzerhof,HSE06) 这一被公认是计算半导体光电性质最为准确的方法,来计算带隙﹑光吸收等光电性质。此外,由于Bi 元素的相对论效应,计算中还考虑了自旋-轨道耦合效应(SOC)。
2 结果与讨论
2.1 完全有序的AgBiS2
如图1 所示,AgBiS2有序结构的原胞是一个菱形六面体,具有空间群结构,其晶格常数为:a=b=c=6.78Å,α=β=γ=34.73°。晶格常数和对称性与实验中合成的结构基本一致[18]。我们进一步计算了这种有序结构的能带结构图和态密度,结果见图2 和图3。能带结构表明AgBiS2是一种间接带隙半导体,价带顶(Valence Band Maximum, VBM)位 于A 点(-0.40, 0.50,0.45),导 带 底(Conduction Band Minimum,CBM) 位 于T 点(-0.50,0.50,0.50)。AgBiS2的基础带隙值 (A →T) 约为0.94eV,与文献报道的AgBiS2纳米晶体带隙的测量值(约1.0eV)[19]基本一致。从计算得到的总态密度和分波态密度图中可以看出,AgBiS2的VBM 主要由Ag 4d﹑S 3p 及Bi 6s 轨道贡献,CBM 主要由Bi 6p﹑Ag 5s 和S 3p 轨道杂化的反键态组成。Ag 4d 和Bi 6s 是非键轨道,不参与成键,却推高了AgBiS2的VBM。图4 显示了计算得到的AgBiS2的光吸收系数。AgBiS2在可见光区域具有超过105cm-1的高吸收系数,并具有间接带隙结构的明显特征,带边吸收(1.46eV)显著高于基础带隙(0.94eV)。
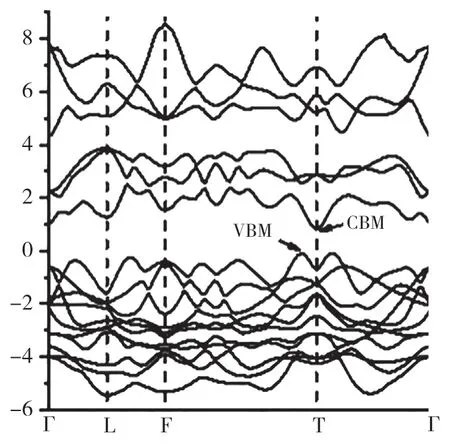
图2 AgBiS2 有序结构的能带结构Fig.2 the band structure of the ordered AgBiS2
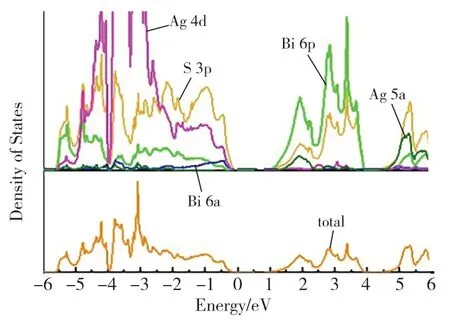
图3 AgBiS2 有序结构的总态密度和分波态密度图Fig.3 the density of statesand of the ordered AgBiS2

图4 AgBiS2 有序结构的光吸收系数Fig.4 the light absorption of the ordered AgBiS2
2.2 完全无序的AgBiS2
与完全有序的AgBiS2结构不同,Ag+和Bi3+这2 种阳离子所占据的位置可能以无序的方式分布,且这种无序度会随着温度的升高而升高。为了描述完全无序的AgBiS2结构,我们采用特殊的准随机结构(SpecialQuasi-RandomStructures,SQS)方法[20]产生了一个小的有限超胞,从而使其平均原子相关函数最接近无序结构的目标原子相关函数,这里的k表示多面体图形中的顶点数,m 表示第m 个相邻距离。使用Ising 模型为每一个位点i 分配一个自旋变量Si, 如果它被Ag+占据,则将其设置为+1,如果被Bi3+占据,则设置为-1。原子相关函数是自旋变量的乘积,对于有序AgBiS2,为-1,而在无序AgBiS2结构中,所有数字均应为0。图5 示意了AgBiS2中Ag 和Bi 原子完全无序原子结构的分布情况,按照这种方法,我们构建了AgBiS2的128 原子立方体超胞准无序结构(SQS),其晶格常数为:a=23Å,b=c=11.5Å,α=β=γ=90°。

图5 AgBiS2 完全无序结构的XY 方向结构示意图Fig.5 The XY direction structure schematic of the fully ordered AgBiS2
我们运用第一性原理的计算方法,对这种结构的光电性质展开了进一步研究。图6 展示了完全无序AgBiS2的能带结构图。可以明显看出,AgBiS2的完全无序结构具有更加直接的带隙。其光学带隙(Γ →Γ)与基础带隙(X →Γ)相差近0.02eV。我们计算了AgBiS2完全无序结构的总态密度和分波态密度,结果见图7。不同于有序结构,完全无序AgBiS2的VBM 主要由Ag 4d﹑S 3p﹑Bi 6s 和Bi 6p 轨道组成,CBM 主要是由Bi 6p 和S 3p 轨道组成。在有序排列时,Bi 6p﹑Ag 5s 轨道主要贡献于CBM 的组成,而在无序结构中,Bi 6p 在VBM 的组成比例大幅度提高,Ag 5s 轨道组成了更高的导带能带,在CBM 几乎无贡献。这种重组后的带边组成成分和能带带隙随有序度的巨大变化,与无序结构局域化学键的畸变直接相关。如图8 所示,对比完全无序结构的光吸收和有序结构的光吸收后可以发现,完全无序相的光学吸收带边,从原来的1.46 eV 红移到0.33eV,从而大范围提升了AgBiS2的光响应范围。同时,根据我们的预测,只要精确控制AgBiS2的有序度,就可以精准控制其光响应范围,使其在0.33~1.46eV 之间连续可调。此外,我们对完全有序结构和完全无序结构的热力学性质进行了计算。计算结果表明,完全有序和完全无序AgBiS2之间的相转变只需0.11eV・(f.u.)-1。 在高温合成的条件下,无序的AgBiS2更容易合成,而在低温条件下,有序的AgBiS2更容易合成。也就是说,仅通过控制合成温度,就可以精确调控AgBiS2的有序度,进而控制其带隙,使其在0.33~1.46eV 范围内变化,从而控制其在可见-红外区的响应范围。
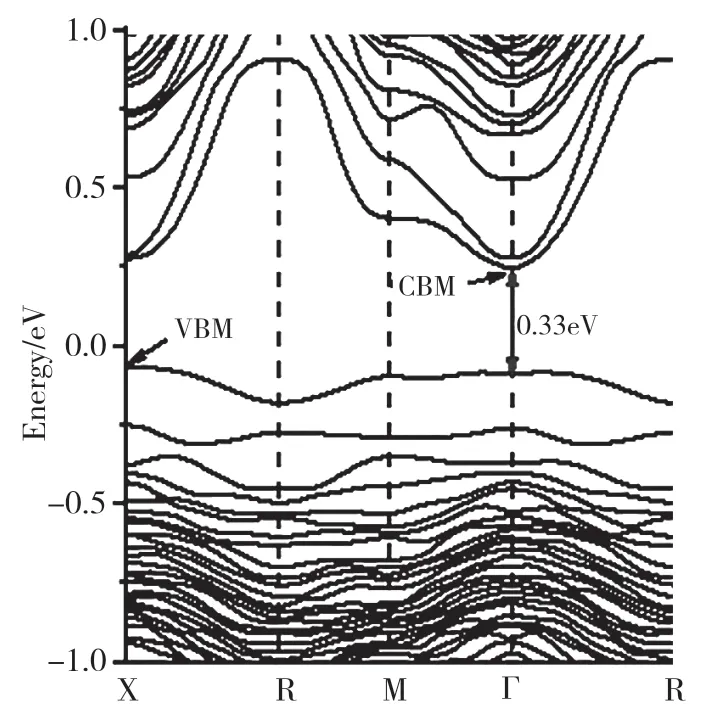
图6 AgBiS2 完全无序结构的能带结构Fig.6 the band structure of the fully ordered AgBiS2
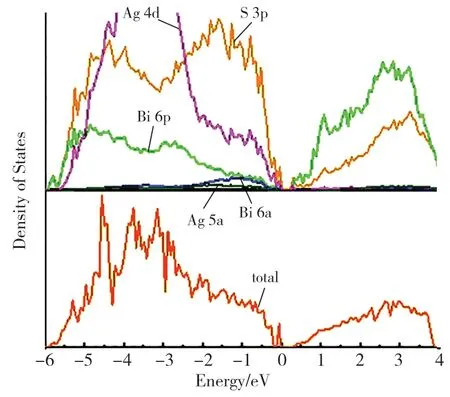
图7 AgBiS2 完全无序结构的总态密度和分波态密度图Fig.7 the density of statesand of the fully ordered AgBiS2
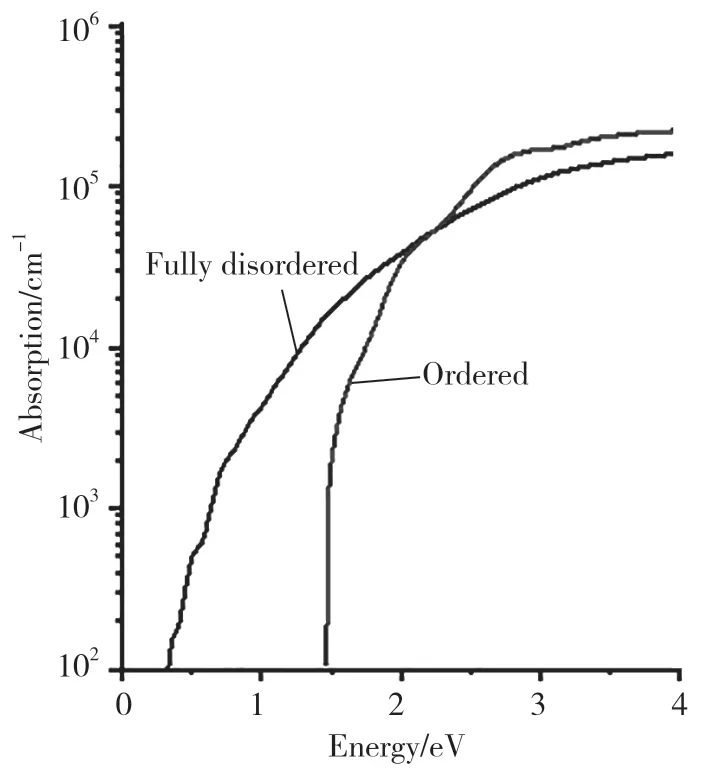
图8 AgBiS2 完全无序结构与有序结构的光吸收对比Fig.8 the light absorption contrast of the ordered AgBiS2 and the fully ordered AgBiS2
3 结论
本文中,我们运用第一性原理的计算方法,结合准无序结构模型,研究了AgBiS2的有序与无序结构的电子结构性质和光学性质,以及从无序到有序的相转变。结果表明,改变AgBiS2结构中Ag+和Bi3+占据的无序度,可以调控AgBiS2的光学带隙在0.33~1.46 eV 范围内连续变化,从而实现近红外和中红外区域的光学响应。此外,从完全无序到完全有序之间的相转变只需0.11 eV・(f. u.)-1,表明通过控制合成温度,可以实现对AgBiS2带隙大小的精准调控,从而控制其红外响应范围。这种组态调控方法为发现和调控新型红外光电材料提供了一条新思路。
猜你喜欢
汽车实用技术(2022年14期)2022-07-30
济南大学学报(自然科学版)(2021年6期)2021-11-10
小天使·聪聪画刊(2021年2期)2021-09-10
汽车零部件(2020年10期)2020-11-09
成都信息工程大学学报(2019年3期)2019-09-25
汉语世界(The World of Chinese)(2019年6期)2019-09-10
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
能源(2017年11期)2017-12-13
山西大同大学学报(自然科学版)(2016年2期)2016-12-12
