
婴儿配方乳粉中VA、VD和VE混合标准物质的研制
2023-08-08 08:59:54宁霄金绍明刘彤彤赵梅曹进
化学分析计量 2023年7期
宁霄,金绍明,刘彤彤,赵梅,曹进
(中国食品药品检定研究院,北京 100050)
维生素A、D、E(VA、VD、VE)均属于脂溶性维生素。这三种维生素对人体均有重要的作用,一但缺乏将导致一些疾病如夜盲、骨质疏松、易感染癌症等[1]。另一方面,该类脂溶性维生素不易通过尿液排出体外,容易在体内大量蓄积,因此VA,VD,VE摄入过量时,可能引起皮肤、骨骼、脑、肝等多种脏器组织病变,对人体健康造成巨大影响[2]。由于婴儿食物来源单一,相关维生素在乳粉中的含量直接影响婴幼儿健康。GB10765—2021《食品安全国家标准 婴儿配方食品》及GB10767—2021《食品安全国家标准 较大婴儿和幼儿配方食品》中均严格限定了这3种维生素的含量范围[3-4]。
婴儿配方乳粉基质复杂,牛乳、脱盐乳清粉、乳清蛋白粉等牛乳分离提取物为主要蛋白原料,复配植物油和乳糖,是脂肪、碳水化合物的原料来源,形成了婴儿配方乳粉的主体原料,同时强化维生素、矿物质等一定量的营养强化剂[5]。产品生产过程中存在分布不均的风险,同时,其稳定性受环境影响较大。据宇盛好等统计的2015~2020 年全国抽检婴幼儿配方乳粉结果[6],这三种维生素指标不合格是存在的主要问题之一,因此,准确测定婴幼儿配方乳粉中VA,VD和VE的含量尤为重要。
为监控实验室对三种维生素的检测能力,保证检测结果有效可靠,且具有溯源性、可比性和准确性,本实验室研制了婴儿配方乳粉中VA,VD和VE含量分析混合标准物质。采用固相萃取法取代国家标准[7]中的有机溶剂萃取法,并使用二维液相色谱法对目标溶液进行分析,省去正相制备步骤,简化样品处理流程,实现一次进样同时完成VA,VD和VE含量的测定,可以满足标准物质定值的要求。该标准物质可用于相关检测实验进行三种组分分析方法的同步确认、过程质量控制及分析仪器校准等工作。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:U3000 型双三元液相色谱(2DLC)系统,配有DAD 检测器,美国赛默飞世尔科技公司。
电子天平:XPE205 型,感量为 0.01 mg,瑞士梅特勒-托利多公司。
维生素专用固相萃取柱:TUP-E76-2060 型,天津诚轴生物科技有限公司。
VA(视黄醇)对照品:纯度(质量分数)不小于98.7%,相对扩展不确定度为 1.77%(k=2),美国西格玛公司。
VD3对照品:纯度(质量分数)不小于99.8%,相对扩展不确定度为 0.3%(k=2),德国DR.E公司。
VE(D-α-生育酚) 对照品:纯度(质量分数)不小于98.2%,相对扩展不确定度为 2.0%(k=2),德国DR.E公司。
无水乙醇、乙腈、甲醇:色谱纯,德国默克公司。
1.2 样品制备
向乳粉企业采购分装前的成品乳粉(生产工艺为湿法喷雾制备,已定量加入维生素A、D、E),混匀,包装。最终用棕色玻璃瓶带硅胶垫塑料盖包装,铝塑袋真空密封,共制备300 瓶,每瓶净含量不少于20 g,保存条件:温度为(25±2)℃,相对湿度为(60±10)%。
1.3 VA、VD和VE含量测定方法
1.3.1 样品预处理
精密称取婴儿配方乳粉5 g 于250 mL 锥形瓶中,加入20 mL 温水(50 ℃)溶解后,加入抗坏血酸1.0 g,BHT(2,6-二叔丁基-4-甲基苯酚)0.1 g,无水乙醇50 mL,质量分数为50%的KOH 溶液15 mL,在65 ℃水浴中震荡皂化50 min。将上述皂化液冷却,用水-乙醇(体积比为1∶1)混合液转移定容至100 mL,摇匀,以8 000 r/min 转速离心5 min,取20 mL上清液,转移至TUP-E76-2060固相萃取柱(20 g/60 mL)中,用100 mL正己烷(含BHT,BHT的质量浓度为 50 mg/L)以约5 mL/min的流量洗脱,将洗脱液收集至旋蒸瓶中,使用旋蒸仪于40 ℃旋蒸至约2 mL,将溶液转移至离心管中,用氮气吹干后准确加入2 mL 甲醇复溶,经0.22 μm 微孔滤膜过滤,作为样品溶液。
1.3.2 定量方法
所制备的乳粉中混合维生素标准物质中VA、VD和VE含量采用液相色谱法测定。流动相为乙腈-甲醇-水;将2D-LC系统的右泵作为一维分析泵,以赛默飞FOODKIT1 ADE-C18色谱柱(150 mm×3.0 mm,3 μm)为一维分析柱,流量为0.5 mL/min;将2D-LC系统的左泵作为二维分析泵,以赛默飞FOCDKIT1& 2ADE-C18色谱柱(100 mm ×4.6 mm,2.6 μm)为二维分析柱,流量为0.8 mL/min;柱温30 ℃;检测波长分别为264 nm(VD)、296 nm(VE)和325 nm(VA);进样体积为20 μL。梯度洗脱条件见表1。

表1 梯度洗脱条件
1.4 均匀性和稳定性检查
根据CNAS—GL 03:《能力验证结果的统计处理和评价指南》中关于均匀性、稳定性检验的要求进行试验及统计分析[8]。
1.4.1 均匀性检查
随机抽取15份样品进行均匀性测定,每份样品做 2 次独立平行测定,对处理得到的待测液按随机顺序进样,采用单因素方差分析和F检验对样品的均匀性进行考察,当测定结果的F值小于Fα(γ1,γ2)时,认为样品为充分均匀的。
1.4.2 稳定性检验
按照先密后疏的原则设置不同的采样时间点,采用同步稳定性评估方法进行稳定性分析。实验温度选择(25±2)℃,相对湿度选择为(60±10)%,进行储存稳定性实验。将样品分别在上述条件下存放,于1、3、6、12个月后分别抽取3份样品, 每份样品检测2次。以均匀性检测结果为长期稳定性0 点,采用回归分析评定样品的稳定性。
1.5 定值
参 考 JJF 1343—2012[9]和ISO GUIDE 35:2017[10],采用同一方法多家实验室联合定值的方式,稳健的统计方法定值。选择8 家行业领域经验丰富且经国家认证的检测实验室或检测中心,分别向每个定值实验室提供样品5 份,每个样品分别平行测定 2 组数据。各定值机构提供 10 组测量数据。回收定值检测结果后,对每一组独立测量结果采用格拉布斯(Grubbs)检验进行离群值检验,采用狄克森(Dixon)检验对组间数据进行离群值检验。剔除离群值后,采用 Kolmogorov-Smirnor(KS)检验对所有数据进行正态分布检验。在符合正态分布的情况下,计算出各实验室检测结果中位值的中位值,该总体中位值即样品的定值结果,同时进行定值的不确定度评定并给出最终结果。
2 结果与讨论
2.1 定量分析条件的优化
由于VA和VE光敏性较强,遇光极易分解,因此婴儿配方乳粉制备工艺中需要通过乙酸酯化并微胶囊化处理,从而减少这三种类脂溶性维生素在运输和储存过程中的流失。在定量分析中,样品需经过皂化处理,才能将VA、VD、VE溶解出来。国家标准方法[11]采用有机溶剂萃取法进一步净化皂化液,由于该方法易出现两相界面乳化现象,导致目标组分在从皂化液转移到萃取液的过程中产生损失,且光敏性强的目标组分在萃取液浓缩过程中极易见光分解,从而该方法回收率较低[12-16]。笔者采用固相萃取柱净化皂化液,减少了复杂操作导致的目标组分损失。考虑到乳粉中VD含量低,为实现与VA、VE的同步检测,需通过大容量的固相萃取柱富集。通过对比,最终选择规格为20 g/60 mL 的TUP-E76-2060 维生素专用固相萃取柱。该固相萃取柱采用特殊工艺处理的硅藻土填充而成,具有最大的比表面积和均衡的表面活性,可给样品提供一个理想的液液分配支撑表面。实验验证结果表明,样品快速通过亲水材料筛板后,水和无机盐等杂质被保留,弱极性物质被正己烷洗脱下来。
2.2 样品的均匀性和稳定性考察
2.2.1 均匀性检验结果
按1.4.1进行均匀性检测,采用单因素方差分析方法对样品测定结果进行数据统计。三种维生素含量测定结果列于表2(“组内1”“组内2”表示两个平行操作样品),F检验分析结果列于表3。结果显示:样品的F计算值小于F临界值,表明样品均匀性良好。
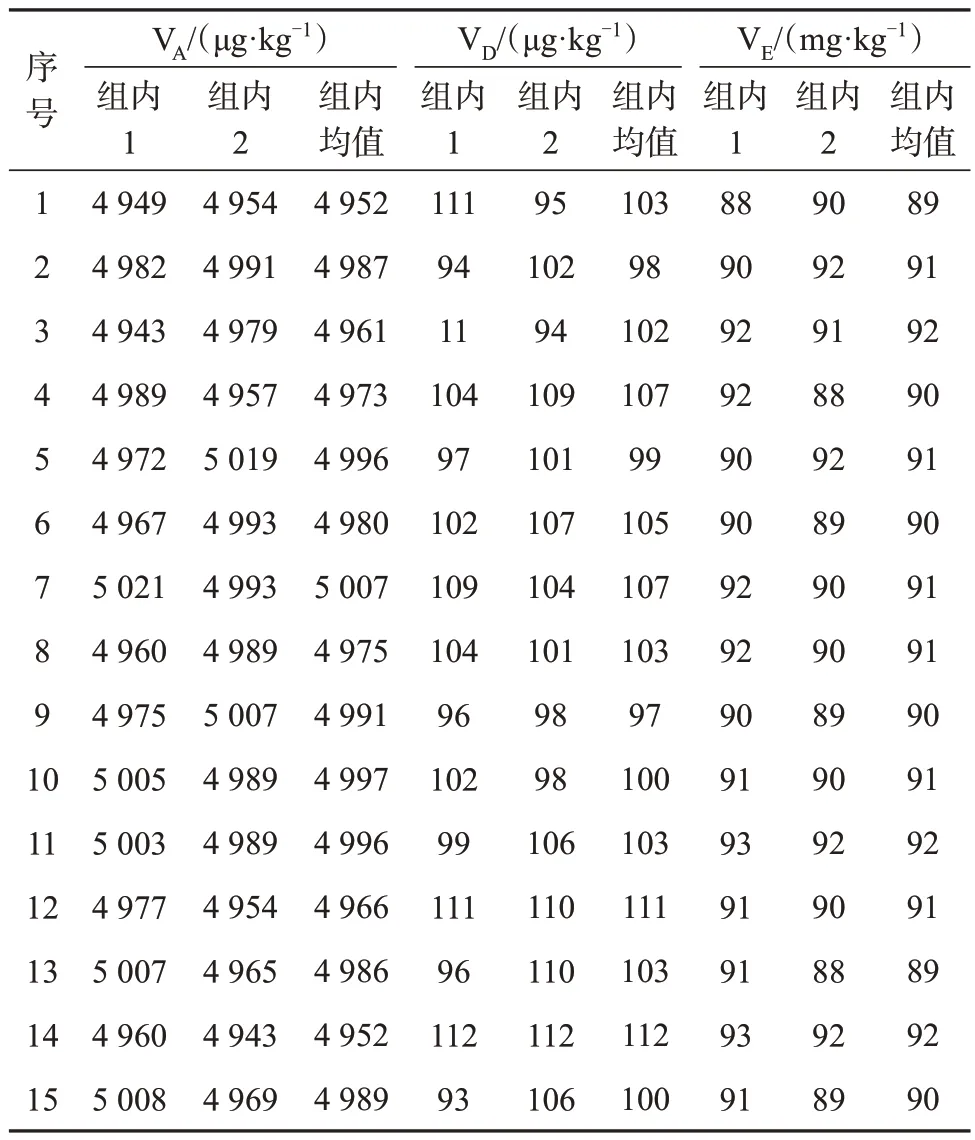
表2 婴儿乳粉样品中三种维生素质量分数测定结果
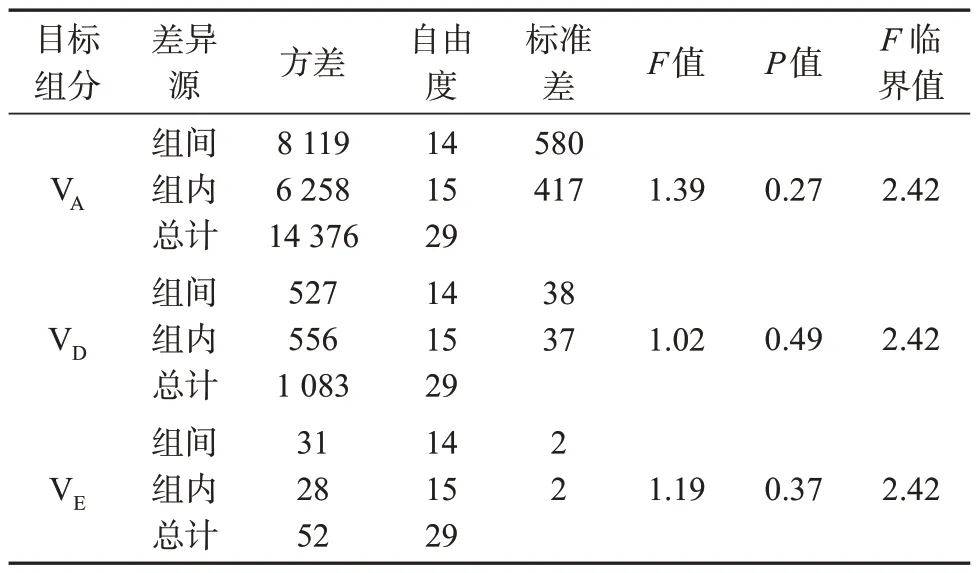
表3 婴儿乳粉样品中三种维生素含量测定方差分析结果
2.2.2 稳定性检验结果
按1.4.2 进行稳定性考察,结果见表4。对表4检测结果以时间-质量分数做线性拟合,并进行回归方差分析,结果见表5。结果表明,在95%的置信水平下,斜率的P>0.05,说明样品在考察期限内稳定性良好。

表5 稳定性检测回归方差分析结果
2.3 定值结果及离群值检验
按照1.5 方法发放协作定值样品,统一采用1.3定量方法,联合8 家行业领域经验丰富且经国家认证的检测实验室协作定值,实验室分别编号为A~H,各协作单位定值检测结果见表6。采用格拉布斯(Grubb’s) 检验,对 8 个实验室内检测结果进行离群值检验。采用狄克森(Dixon) 检验对实验室间检验结果的一致性进行评价。在95% 的置信水平,均无离群值。计算各协作定值单位结果的中位值,并对各协作定值单位中位值结果做二次中位值计算,作为指定值。最终VA、VD和VE含量定值结果分别为4 987 μg/kg、104 μg/kg和90 mg/kg。
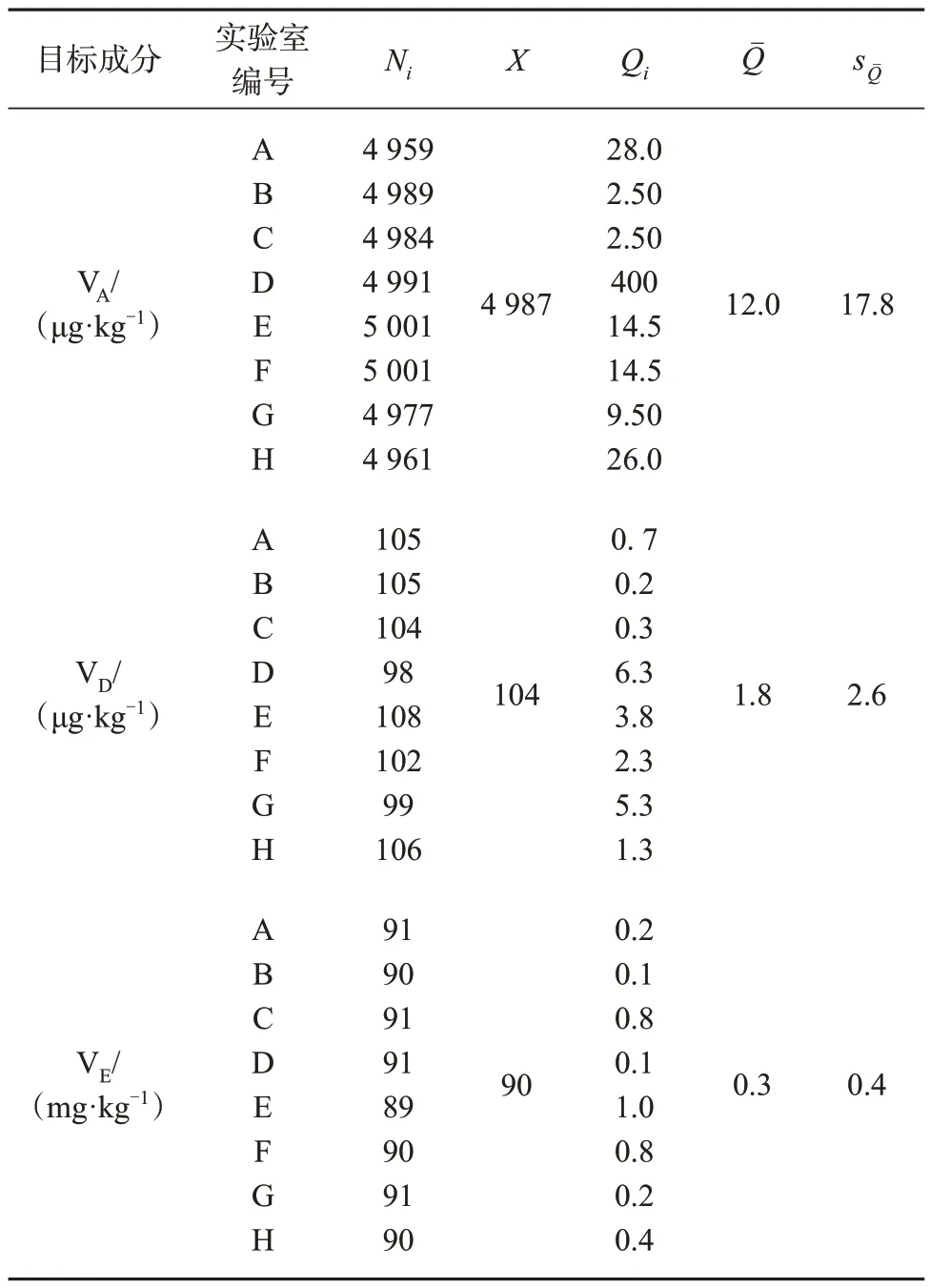
表6 婴儿配方乳粉中三种维生素协作定值结果
2.4 定值不确定度的评定
2.4.1 样品协作定值引入的不确定度
计算各协作定值单位结果的中位值,记为Ni。以这些中位值的中位值作为指定值,记为X。计算N1,N2…Ni与X之差的绝对值,记为Qi。Q1,Q2…Qi的中位值记为Qˉ。各定值实验室结果及统计见表6。协作定值的不确定度按式(1)计算,结果见表6。
式中:uD——协作定值的标准不确定度;
n——协作标定的实验室数量;
sQˉ——正态分布的标准偏差,sQˉ=Qˉ/0.674。
协作定值的相对标准不确定度按式(2)计算,结果见表8。
式中:uD,rel——协作定值的相对标准不确定度;
X——各实验室报告值的中位值。
2.4.2 样品均匀性引入的不确定度
均匀性引入的不确定度及相对不确定度分别按式(3)、式(4)计算,结果见表7。

表7 定值的标准不确定度计算结果
式中:ubb——均匀性引入的标准不确定度;
n——每份样品平行测定的次数;
ems-in——组内的均方差;
vin——组内自由度。
式中:ubb,rel——均匀性引入的相对标准不确定度;
m——均匀性检验数据的平均值。
2.4.3 样品稳定性引入的不确定度
将样品稳定性检验结果按时间–质量分数做线性拟合,所得拟合直线的残差为斜率不确定度,如式(5)。按式(6)、式(7)分别计算样品稳定性引入的标准不确定度和相对标准不确定度,结果见表7。
式中:s(b1)——斜率b1的不确定度;
sˉ——回归直线残留平均标准偏差。
式中:ults——样品稳定性引入的标准不确定度;
s(b1)——样品稳定性考察直线拟合斜率的标准不确定度;
t——样品的有效期(稳定性考察期)。
式中:ults,rel——样品稳定性的相对标准不确定度;
m——所有稳定性检验测定结果的平均值。
2.4.4 合成不确定度
以定值、均匀性、稳定性引入的不确定度分量合成标准物质定值的相对标准不确定度,按式(8)计算。按式(9)计算标准物质的合成标准不确定度,结果见表7。
按95%的置信区间,取k=2,按式(10)计算标准物质的扩展不确定度,VA、VD和VE结果分别为3.10、1.59和1.30。
式中:uCRM——标准物质的标准不确定度;
UCRM——标准物质的扩展不确定度;
k——包含因子。
2.5 婴儿配方乳粉标准物质的定值结果
经过均匀性检验,12 个月内的3 个月次的稳定性检验和8 家实验室协作定值,最终确定婴儿配方乳粉标准物质中VA、VD和VE含量定值结果及其扩展 不 确 定 度(k=2) 分 别 为(4987±3.1) μg/kg、(104±1.59) μg/kg和(90±1.3) mg/kg。
3 结语
通过优化样品处理及检测方法,建立了婴儿配方乳粉中VA、VD和VE含量同步分析标准物质的二维液相色谱定值方法,该方法利用固相萃取方式,省去了有机溶剂液液萃取的繁杂步骤,实现了三种脂溶性维生素的同步检测,在提升方法的准确率及灵敏度的基础上,提高了效率,适用于大批量样品的同时测定。用此法检验标准物质的均匀性和稳定性,均满足要求;且多家实验室联合定值方式,也再次验证该方法具备重现性、可靠性和准确性。通过该方法制备的婴儿配方乳粉基体标准物质可用于日常食品质量控制、检测方法的开发及实验室能力验证等领域,也为其他基体标准物质的制备提供了参考。
猜你喜欢
食品科学(2023年4期)2023-03-06 12:49:32
中学生数理化(高中版.高二数学)(2022年1期)2022-04-26 13:59:54
新世纪智能(教师)(2021年2期)2021-11-05 08:43:26
食品安全导刊(2021年21期)2021-08-30 08:21:58
河北理科教学研究(2020年3期)2021-01-04 01:49:52
中国乳业(2020年12期)2020-04-12 01:12:46
小学科学(学生版)(2020年2期)2020-03-03 13:40:18
小学科学(学生版)(2020年1期)2020-01-19 06:02:10
电子制作(2018年10期)2018-08-04 03:25:02
电子制作(2018年12期)2018-08-01 00:48:08
