
咖啡酸国家一级纯度标准物质的研制*
2023-08-01 09:26苏斌张宝喜毕研才龚宁波杨世颖张丽吕扬杜冠华
医药导报 2023年8期
苏斌,张宝喜,毕研才,龚宁波,杨世颖,张丽,吕扬,杜冠华
(1.山东济世药业有限公司质量部,济南 271100;2.北京协和医学院·中国医学科学院药物研究所晶型药物研究北京市重点实验室,北京 100050;3.北京协和医学院·中国医学科学院药物研究所药物靶点研究与新药筛选北京市重点实验室,北京 100050)
咖啡酸属酚酸类化合物,广泛存在于多种中药材植物中,如一枝黄、杜仲、薄荷、蒲公英等。研究发现,咖啡酸具有广泛的药理活性,如抗菌、抗病毒、抗炎、抗氧化、抗癌等作用,近年来又逐渐发现其在糖尿病肾病、心肌细胞保护、免疫系统疾病等方面具有较好活性[1-5]。鉴于咖啡酸化学成分存在于多种中药材中,《中华人民共和国药典》将其列为冬葵果等中药材质量评价的有效或指标成分,在冬葵果、杠板归、牵牛子等多种中药材的鉴别检查中亦作为对照品使用[6]。因此,研究咖啡酸纯度标准物质对于我国中药相关产品的质量控制具有重要的科学意义。
标准物质是具有可溯源性和准确量值的测量标准,其基本功能是可以复现、保存与传递量值,保证在不同时间与空间中量值的可比性与一致性。标准物质的特性量值是作为分析测量中的“量具”和“标准”,在校准仪器、评价方法、确定材料成分、考核人员水平以及生产过程中的质量控制方面起着重要的标准的作用[7-8]。2005年国际标准化组织/标准物质委员会(ISO/REMCO)对标准物质、有证标准物质、基准标准物质赋予了新的定义[9-10]。
我们根据中华人民共和国计量法、一级标准物质研制技术规范(JJF 1006-1994)、标准物质管理办法与《中华人民共和国药典》等相关技术文件[11-14],研制了咖啡酸化学纯度有证标准物质,具有溯源性和量值传递功能,可应用于相关化学药品、中药产品、保健品的质量监控,为实现我国准确定量检测药品与相关物质提供了标准物质资源和物质纯度标准。
根据有证标准物质研制要求,笔者在本实验完成了咖啡酸纯度标准物质的均匀性检验、短期稳定性考察、长期稳定性考察等系列研究,并采用质量平衡法与库仑滴定法两种不同原理的检测分析技术,实现了对咖啡酸化学纯度标准物质联合定值,建立了不同技术方法的不确定度评估数学模型,确保了标准物质定值结果的科学性和准确性。
1 仪器与试剂
1.1仪器 Agilent 1200 型高效液相色谱仪(配置:四元梯度泵,自动进样器,DAD检测器,Agilent chemstation色谱工作站);库仑滴定仪(自制,双铂电极,电流指示器);XS 105型分析用天平(METTLER TOLEDO公司,感量:0.01 mg);TU-1901 型双光束紫外可见分光光度计(北京普析通用仪器有限责任公司)。研究使用的高效液相色谱仪、紫外分光光度计、分析用天平等仪器及玻璃器皿、移液枪等均经国家计量院校准,库仑滴定仪经国家一级亚砷酸根溶液标准物质(GBW 08666)校准。
1.2试剂 乙腈(色谱纯,Fisher Chemical);异丙醇(色谱纯,Fisher Chemical);冰醋酸(分析纯,北京化学试剂公司);溴化钾(分析纯,北京化学试剂公司);盐酸(分析纯,北京化学试剂公司);纯水;流动相使用前均过孔径0.2 μm微孔滤膜。
2 方法与结果
2.1高纯度标准物质候选物制备 咖啡酸标准物质候选物原料由东北产地的蒲公英中药材提取、分离、纯化获得。 采用四大光谱技术进行咖啡酸纯度标准候选物样品进行结构确证研究:UV(乙腈)λmax:320.5 nm、294.0 nm、239.0 nm、216.5 nm、191.5 nm;IR:3 431.7,3 231.7,3 025.0,2 830.4,2 563.9,1 644.7,1 619.4,1 600.7,1 530.7,1 449.6,1 374.7,1 352.0,1 326.1,1 296.1,1 280.0,1 217.7,1 174.3,1 120.3,974.8,968.6,935.4,900.2,871.9,849.7,816.8,801.7,779.9,736.6,699.1,647.8,603.0,576.0,458.6 cm-1;H-NMR(d6-DMSO):δppm:6.2(1H,d,J=15.9Hz,H-6),6.7(1H,d,J=8.2Hz,H-7),6.9(1H,dd,J=8.2Hz,J=1.85Hz,H-2),7.0(1H,d,J=1.85Hz,H-8),7.4(1H,d,J=15.85Hz,H-5),9.1(1H,s,H-1),9.5(1H,s,H-3),12.1(1H,s,O-H)。13C-NMR(d6-DMSO):δppm:114.6(C-1),115.1(C-6),115.7(C-7),121.1(C-5),125.7(C-2),144.6(C-4),145.5(C-3),148.1(C-8),167.9(C-9);ESI-MSm/z:181.0(M++1);以上数据与文献报道基本一致[15],可确定标准物质候选物样品为咖啡酸。
2.2定值方法的方法学研究 咖啡酸纯度标准物质采用质量平衡法与库仑滴定法2种不同原理的方法进行联合定值。其中质量平衡法是首先采用高效液相色谱面积归一化法,测定候选物中主成分含量,再扣除候选物中无紫外吸收的杂质成分含量,从而获得待测标准物质候选物的化学纯度。
2.2.1高效液相色谱法研究
(1)色谱条件 色谱柱:Agilent Eclipse XDB C18(150 mm×4.6 mm,5 μm)。流动相:梯度洗脱,A相:0.5%冰醋酸溶液(含5%异丙醇);B相:乙腈。5%Bisocratic (9 min);5%~25%B linear(5% min-1,4 min);25%B isocratic(9 min);25%~5%B linear(5% min-1,4 min);5%B isocratic(9 min)。流速:1.0 mL·min-1,柱温:35 ℃,进样量:10 μL。有关物质检测浓度:500 μg·mL-1。记录色谱图及峰面积,利用Agilent Chemstation色谱工作站处理数据,采用面积归一化法进行纯度测定。
(2)线性范围 精密称取咖啡酸纯度标准物质样品25.0 mg,10%乙腈溶解并定容至25 mL量瓶,摇匀,配制成浓度为1 000 μg· mL-1的储备液。分别精密量取储备液0.1,0.5,1.0,2.5,5.0,7.0 mL,用10%乙腈定容至10 mL容量瓶中,配制成系列浓度分别为10.0,50.0,100.0,250.0,500.0,700.0 μg·mL-1的标准溶液。分别精密度量取各浓度溶液10 μL注入液相色谱仪,记录色谱图及峰面积。按“2.1.1”项下色谱条件进样分析。以样品浓度(X,μg·mL-1)为横坐标,以峰面积(Y)为纵坐标,绘制标准曲线,回归线性方程为Y=32.556X+33.788,相关系数R2=0.999 9(n=6)。结果表明,在10.0~700.3 μg·mL-1浓度范围内,咖啡酸峰面积与浓度呈现良好的线性关系。
(3)仪器精密度 精密称取咖啡酸纯度标准物质12.5 mg,加10%乙腈溶解,并定容至25 mL量瓶中,配制成浓度为500.0 μg·mL-1的溶液,摇匀,备用。精密量取溶液10 μL注入液相色谱仪,重复进样6次。计算6次重复实验的平均峰面积RSD值为0.04%,<2.0%,表明利用高效液相色谱法测定咖啡酸纯度标准物质的检测分析仪器精密度良好。
(4)方法精密度 按“2.1.3”项下方法配制供试品,共6份。精密量溶液10 μL注入液相色谱仪,重复进样3次。计算获得6份样品平均峰面积的RSD值为0.15%,<2.0%,表明利用高效液相色谱法测定咖啡酸纯度标准物质的检测分析方法具有良好的精密度。
2.2.2库仑滴定法
(1)反应原理 电极材料:双铂电极;发生电极反应:2Br—=Br2+2e;电解电流:0.9872 mA;指示终点:电流上升法,电流超过20 μA停止电解;电解质组成:8N盐酸:2M 溴化钾:冰醋酸=1:1:2。咖啡酸分子量为180.15。反应原理见图1。
(2)线性范围 精密称取10.28,12.43,14.11,16.91,18.31,20.91 mg,置于10 mL量瓶中,使用甲醇定容,待用。分别移取100 μL的供试溶液样品进行实验。利用咖啡酸样品量与溴库仑滴定反应时间之间的定量关系进行测定,记录反应时间。以咖啡酸纯度标准物质的进样量为横坐标,以平均反应时间为纵坐标,绘制标准曲线,回归线性方程为:Y=1 078.8X+0.541 2,相关系数R2为0.999 8(n=6)。结果表明,在0.10~0.21 mg质量范围内,物质进样量与反应时间呈现良好的线性关系。
(3)仪器精密度 精密称取15.01 mg,置于10 mL量瓶中,加入甲醇溶解并定容。进样体积为100 μL,重复测定6次,记录反应时间。计算6次重复实验的的反应时间的RSD值为0.42%,表明利用库仑滴定法测定咖啡酸纯度标准物质的检测分析仪器精密度良好。
(4)方法精密度 按“2.2.3”项下方法配制供试品,共6份。进样量为100 μL,记录反应时间。计算相同重量的6份样品的平均反应时间的RSD为0.18%,<2.0%,表明采用库仑滴定法咖啡酸纯度标准物质的检测分析方法具有良好的精密度。
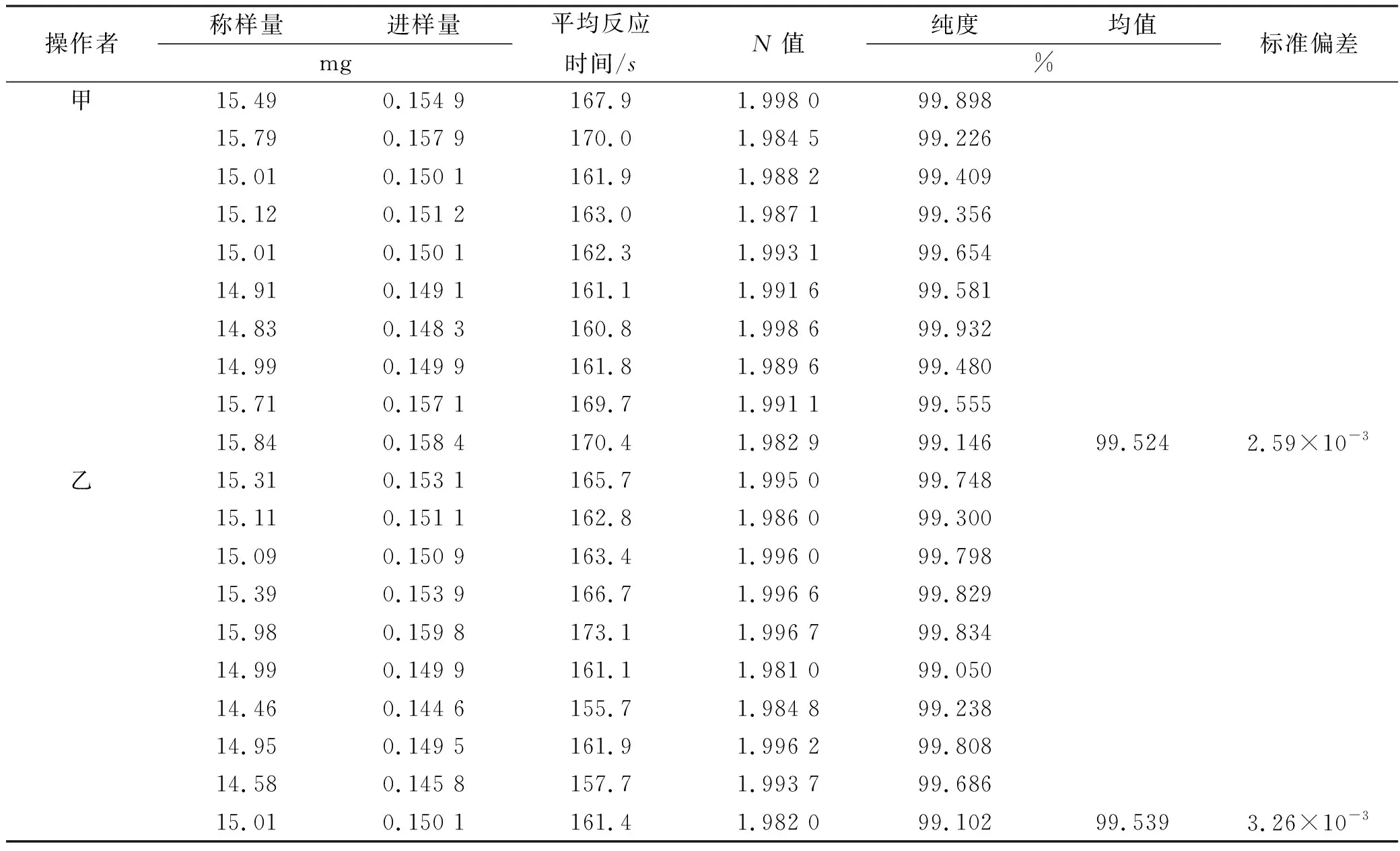
2.3均匀性检验 从500支咖啡酸纯度标准物质最小包装中随机数字表法抽取15 支样品,每支平行取样3 次,加10%乙腈配制成500 μg·mL-1的溶液,按照“2.1.1”项下色谱条件进行均匀性检验,采用HPLC 面积归一化法进行测定,获得45个均匀性检验数据,结果见表1。
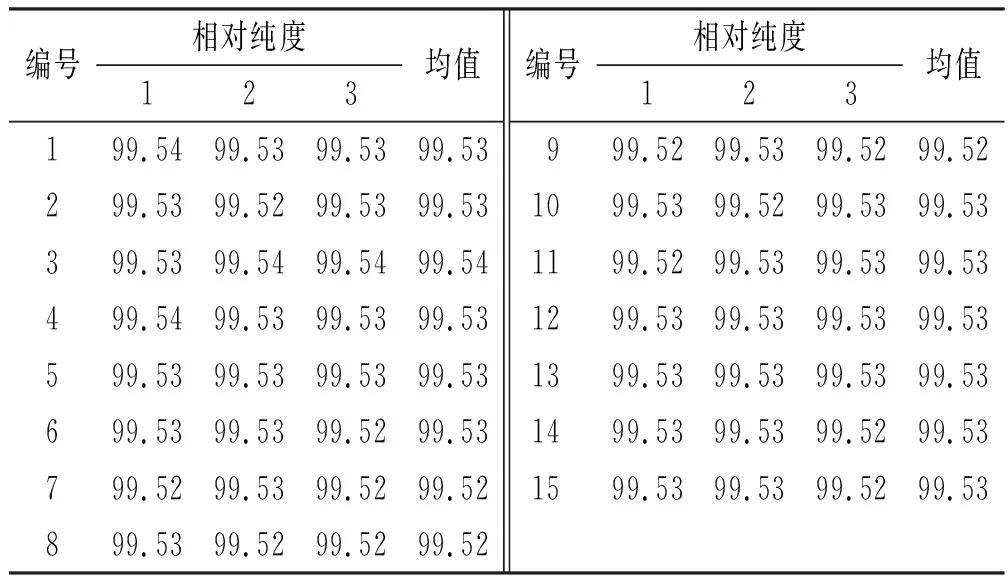
表1 均匀性检验测定结果
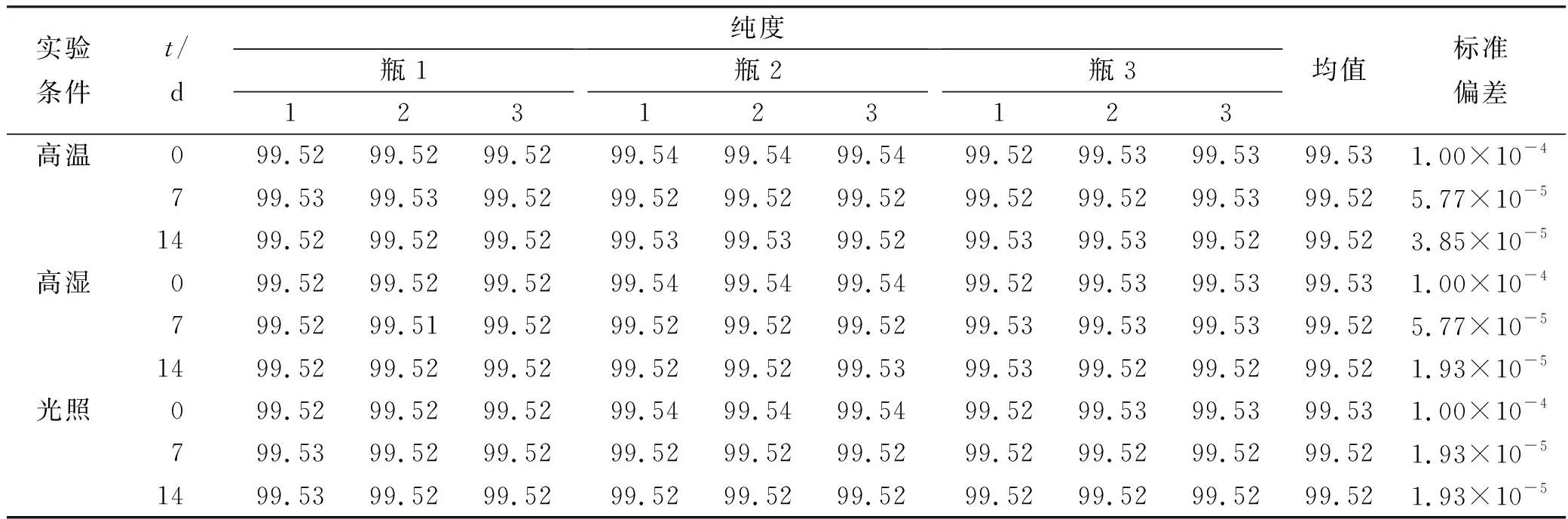
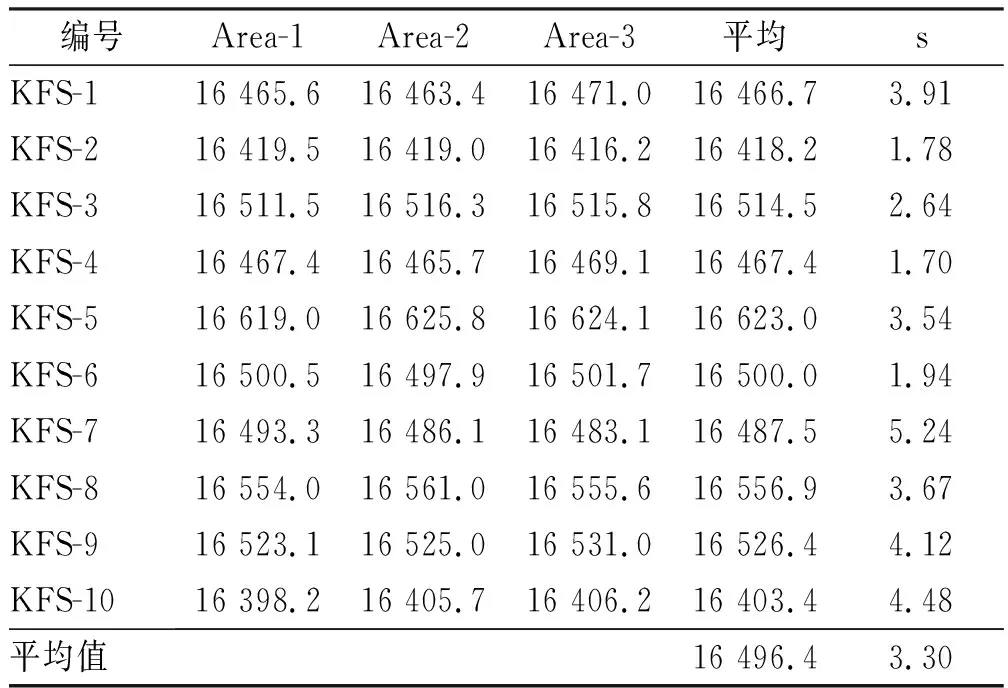
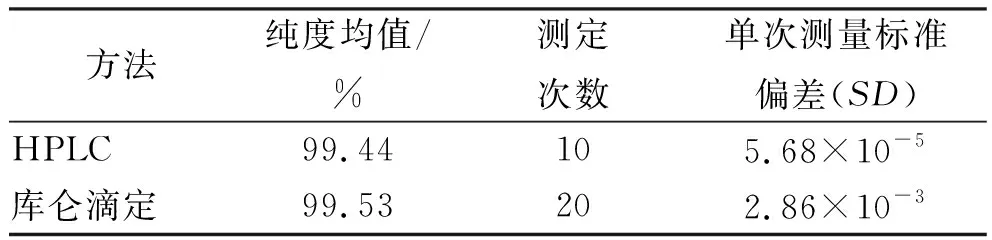
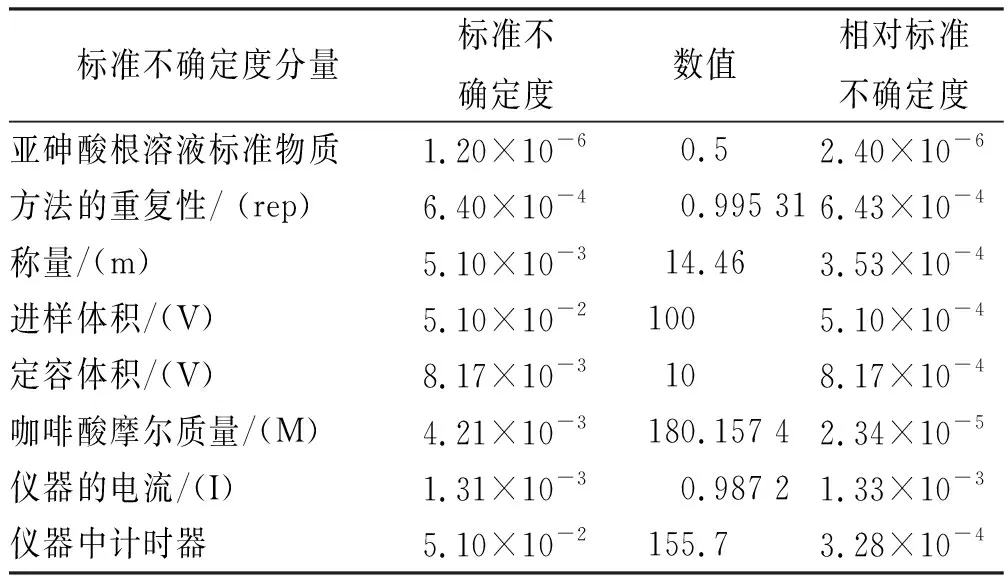
采用单因素方差分析对均匀性检验数据进行统计分析,F=1.76;查F检验临界值表知:F0.05(14,30)=2.04,F 表2 方差分析结果 2.4稳定性考察 2.4.1短期稳定性考察 对包装后标准物质样品进行高温实验(60 ℃)、高湿实验[(90±5)%,25 ℃]、光照实验[照度(4 500±500) lx],并分别于0,7,14 d对经过高温、高湿、光照后的标准物质样品进行检测,每个时间点随机抽样3瓶样品,每瓶进3针,采用HPLC法检测样品纯度,结果见表3。 表3 短期稳定性实验结果 标准物质特性量值的标准值未知时,用平均值一致性检验法评价标准物质的稳定性,公式表述为: 短期实验结果表明,经上述3种影响因素实验前后标准物质样品的化学纯度未发生变化,包装标准物质样品在短期不同环境条件下的稳定性良好。 2.4.2长期稳定性 将包装后标准物质样品放置在常温下长期保存,并分别于0,1,2,4,6,12个月对取样进行纯度检测。每个时间点随机抽样6瓶样品,按短期稳定性考察方法检测样品纯度。结果见表4。 表4 长期稳定性研究数据 2.5标准值的确定 2.5.1质量平衡法 按“2.1.3”项下方法配制供试品,共10份。按上述色谱条件进行测定,记录色谱峰面积,并计算其相对纯度。检测结果见表5和表6。采用HPLC面积归一化法测定标准物质主成分化学纯度的计算数学模型为:WHPLC=100%×A化合物色谱峰面积/A总色谱峰面积,其中WHPLC表示化合物的相对纯度,A化合物色谱峰面积表示测定被测化合物色谱峰面积值,A总色谱峰面积表示测量化合物加各杂质成分的色谱峰面积之和。首先,对10 组测量纯度数据进行格拉布斯(Grubbs) 检验以剔除可疑值,计算得G=1.585,查格拉布斯临界值表知:G0.95(10)=2.176。因G 表5 咖啡酸主色谱峰面积结果 表6 总色谱峰面积 由表5的定值数据的化合物色谱峰面积确定咖啡酸纯度标准物质主成分的纯度为99.53%。鉴于高效液相色谱面积归一化法测定纯度值的局限性,根据质量平衡法原理,需要扣除样品中在高效液相色谱检定波长下不出峰的水分、炽灼残渣和溶剂残留的成分含量。分别采用干燥失重法、炽灼残渣法、气相色谱法测定相关物质的含量。实验结果表明,样品中水分含量为0.030%,炽灼残渣为0.053%,溶剂残留为0.004%,合计0.087%。 最终可得咖啡酸纯度标准物质的纯度标准值为99.53%×(100%-0.087%)=99.44%。 2.5.2库仑滴定法 按随机数表抽取20支标准物质样品,采用甲乙双人独立操作,精密称取约15.00 mg,置于10 mL量瓶中,使用甲醇定容,待用。利用体积计算含咖啡酸样品量,采用100 μL进样量进行实验,记录反应时间。库仑滴定法的数学模型为:x=(W/W样)×100% =[(Q×M)/(n×F)/(mV1/V2)]×100% =[(i×t×M)/(n×F)/(mV1/V2)]×rep×100%,式中W表示反应物质的量 (g);W样表示反应样品进样量 (g);V1表示实验进样量体积 (μL);V2表示容量瓶体积 (μL);Q表示通过电极的电量(C);i表示电流(A);t表示时间(s);M表示反应物质分子量;n表示电子数;F表示法拉第常数 (96 486.57库仑/当量),测定结果见表7。对得到的20组咖啡酸化学纯度标准物质纯度值采用格拉布斯检验进行可疑值的剔除,经计算得G甲=1.576,G乙=1.504。查格拉布斯临界值表知:G0.95(10)=2.176。因G值均小于G0.95(10),故无可疑值存在。2次独立检测数据的t检验结果为0.12,小于临界值2.10,表明两次独立检测数据无显著性差异。 表7 咖啡酸化学纯度标准物质的库仑滴定定值检测结果 库仑滴定法确定的咖啡酸标准物质的化学纯度标准值为99.531%。 表8 咖啡酸纯度标准物质两种原理方法定值结果 2.6不确定度评定 2.6.1定值引入的不确定度定度分量分别为:u(A主色谱峰)=3.344 1;u(A总色谱峰)=3.501 8。 由“5.1”项下测定结果可知,咖啡酸化学纯度标准物质中的水分、无机杂质与溶剂残留的含量测定值均小于 0.1%,对定值结果影响较小,故由其引入的不确定度可以忽略不计。 计算质量平衡法测量的合成不确定度0.002 93。 (2)库仑滴定法定值测量引入的不确定度 由纯度计算数学模型可知,库仑滴定法引入的不确定度评定分量见表9。 表9 库仑滴定法的合成标准不确定度评定分量 根据表7数据计算获得由库仑法试验引入的各种不确定度分量值,详细结果见表9中相对标准不确定度列数据。计算库仑滴定法测量的合成不确定度1.83×10-3。 由表1计算获得样品均匀性引入不确定度值为2.23 ×10-9,因均匀性产生的不确定度较小,可以忽略。 2.6.3样品稳定性引入不确定度 根据样品长期稳定性考察数学模型,斜率的不确定度为: 由表4计算获得样品稳定性引入的不确定度值St=12×Sa=3.348×10-5,由此值可知,因长期稳定性产生的不确定度较小,可以忽略。 2.6.4合成不确定度计算 故总合成不确定度仅由定值方法不确定度引入,计算获得总合成不确定度值为3.45×10-3。 2.6.5扩展不确定度计算 数学模型为U=kuc(总),式中U表示扩展不确定度;k表示包含因子;uc(总) 表示总合成不确定度。k取 2,置信水平P为95%,计算扩展不确定度值为0.69%。 2.7定值结果及不确定度表示 咖啡酸纯度标准物质的纯度值为(99.5±0.7)%,k=2,P=0.95。 论文首次采用2 种质量平衡法和库仑滴定法不同原理技术方法对咖啡酸纯度标准物质进行联合定值研究,有效克服了仅采用单一原理技术方法定值带来的分析方法缺陷和不足,使纯度标准物质定值结果更加准确可靠,为药品相关纯度标准物质的研制提供了技术方法参考。 本文研制的咖啡酸纯度标准物质具有准确的纯度标准值和不确定度,在国内外属首次研制,并已经获得国家质量监督检验检疫总局批准为国家一级计量有证标准物质,证书编号为GBW09529。该标准物质具备量值溯源和传递功能,不仅可用于相关中药材、中成药、提取物、药物中间体等产品的质量控制,还可用于仪器校准、新方法确认等研究,具有非常广泛的实际应用价值。
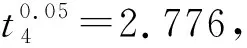



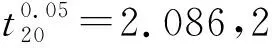
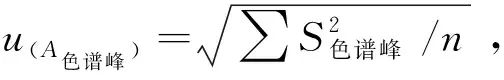
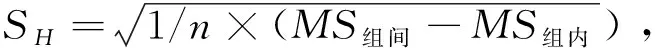
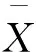

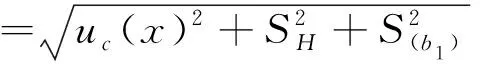
3 讨论
猜你喜欢
中学生数理化(高中版.高二数学)(2022年1期)2022-04-26云南化工(2021年11期)2022-01-12食品安全导刊(2021年20期)2021-11-28新世纪智能(教师)(2021年2期)2021-11-05地震研究(2021年1期)2021-04-13中学生数理化(高中版.高考理化)(2020年3期)2020-05-30电子制作(2018年10期)2018-08-04电子制作(2018年12期)2018-08-01河北地质(2016年2期)2016-03-20中国地震(2015年1期)2015-11-08
