
EGFR酪氨酸激酶及其抑制剂的研究进展
2023-07-21 01:15朱美灵夏德斌
黑龙江大学自然科学学报 2023年3期
关 静, 朱美灵, 夏德斌
(1.哈尔滨医科大学 药学院, 哈尔滨 150081; 2.哈尔滨工业大学 化工与化学学院, 哈尔滨 150001)
0 引 言
EGFR是原癌基因c-erbB1的表达产物,是人表皮生长因子受体(Human epidermal growth factor receptor, HER)家族成员之一。研究发现,EGFR分为三区:胞外配体结合区、跨膜区和胞内激酶区,广泛分布于各种上皮细胞的细胞膜上。EGFR基因位于第七号染色体上,由118个碱基组成,包括28个外显子。其转录形成的mRNA长约5.6 kb,编码的EGFR是分子量为170 kD的跨膜糖蛋白。编码蛋白由1 186个氨基酸组成,具有氨基酸激酶(Tyrosine kinase, TK)活性,可以将胞内信号传递到胞外[1]。已报道的EGFR配体有表皮生长因子EGF、结合肝素的EGF、转化生长因子TGF、β-细胞素和双调蛋白等[2]。EGFR信号通路过度活化和EGFR自身过度表达是恶性肿瘤的主要发生机制[3-4]。EGFR相关信号通路主要有RAS/MAPK、NF-KB、PI3K/AKT/mTOR和JAK/STAT等[5]。与配体结合后,EGFR由单体转化为二聚体,然后激活它位于细胞内的激酶通路,包括Y992、Y1045和Y1148等激活位点,使下游磷酸化,诱导细胞增殖,肿瘤生长。而在实体肿瘤中EGFR高表达或异常表达,研究表明,EGFR基因突变和蛋白的过度表达都可以激活下游信号通路[6],这与癌症密切相关。
主要的EGFR靶向药物可以分为两大类:一类是单克隆抗体(简称单抗),作用于胞外区,竞争性地抑制配体与EGFR的结合,从而阻断EGFR酪氨酸激酶的活化;另一类是小分子酪氨酸激酶抑制剂(Tyrosine kinase inhibitors, TKIs),它进入细胞内,干扰三磷酸腺苷(Adenosine-triphosphate, ATP)结合,抑制21 bp氨基酸激酶磷酸化。虽然EGFR靶向治疗已成功应用于临床并取得了一定的疗效,但治疗过程中患者出现的耐药现象(包括原发性耐药和获得性耐药两种)仍是一个亟待攻克的难题。发展和创新EGFR抑制剂是一个重要的研究目标,本文对TKIs代表药物进行综述。
1 EGFR-TKIs的研发进展
恶性肿瘤是一类严重危害人类健康的疾病,研究和开发高效、选择性强的低毒抗肿瘤药物是药学工作者的目标之一。肺癌是世界上最常见的癌症,每年都有超过100万人死于肺癌,而80%~90%患者为非小细胞肺癌(Non small-cell lung cancer, NSCLC)[7]。在非小细胞肺癌中,EGFR突变大约占12%~47%,其中最为常见的两种突变是19号外显子的缺失突变(del19)和21号外显子的点突变(主要是L858R)[8]。EGFR是癌症生长的刺激因素,与肿瘤的发生有密切关系,EGFR-TKIs成为抗肿瘤药物研发的重要方向。
吉非替尼(Gefitinib, ZD1839)和厄洛替尼(Erlotinib, OSI774)是第一批用于治疗非小细胞肺癌的EGFR-TKIs,并取得一定疗效,两者对野生型EGFR有效,对L858R和delE746-A750有敏感性[9]。Gefitinib是一种口服型、苯胺喹唑啉类可逆性酪氨酸激酶抑制剂。吉非替尼在联合化疗中取得明显效果,它在与顺铂、卡铂、奥沙利铂和紫杉醇等细胞毒性药物联合用药时能增强抑制作用,促进肿瘤细胞凋亡[10]。厄洛替尼(Eelotiinib, OSI774)是第一个获得美国食品公司药品管理局批准的一类口服型EGFR-TKIs药物,与化疗类药物联合使用可以获得更好的治疗效果和生存期,改善患者生存质量[11]。然而,患者对这些抑制剂在9~13个月后产生获得性耐药。常见的与耐药相关的EGFR突变有19号外显子缺失、L858R突变和T790M突变,而大约50%~60%的耐药是由于EGFR T790M突变[12]。另外,埃克替尼(Icotinib, BPI-2009)于2011年6月7日获中国食品药品监督管理总局批准上市,打破了非小细胞肺癌,特别是晚期非小细胞肺癌治疗由进口药品垄断的格局。埃克替尼对晚期NSCLC的疗效与吉非替尼相当,安全性较吉非替尼更优,中位无进展生存期为4.6个月。TKIs靶向药物对血脑屏障的穿透率分别为埃克替尼 6.1%,厄洛替尼2%~4%,吉非替尼1%,因此对于存在脑转移和21号外显子突变的患者,埃克替尼是第一代TKIs药物中的首选用药。
德国勃林格殷格翰公司研发的第二代EGFR-TKIs药物阿法替尼(Afatinib, BIBW2992)是一种新型口服制剂,是EGFR/HER2双重酪氨酸激酶受体的不可逆抑制剂。它与具有催化作用的EGFR第773位酪氨酸结合,以非ATP竞争方式使受体蛋白不可逆改变,对EGFR野生型和突变型包括T790M都有很好的抑制作用[12]。阿法替尼与T790M突变的结合能力是第一代TKIs的100倍以上。阿法替尼与西妥昔单抗联合治疗可有效抑制T790M耐药突变的NSCLC原代细胞[14]。针对EGFR激活突变的患者,通过阿法替尼和顺铂分别联用培美曲塞的对比,可观察到阿法替尼与培美曲塞联用对T790M突变的NSCLC存在明显的协同生长抑制作用,尤其是对T790M突变可逆性EGTR-TKIs耐药患者[15]。但是阿法替尼对野生型EGFR和突变型EGFRs有相同的结合力,缺失了对野生型EGFR的选择性,导致产生毒性和较严重的副作用[16]。达克替尼(Dacomitinib, PF299804)是美国辉瑞公司研制的第二代、不可逆的EGFR酪氨酸激酶抑制剂,该药作用机制类似阿法替尼,能不可逆抑制三种不同ERBB家族分子成员,包括EGFR(HER1)、HER2和HER4。达克替尼用于携带EGFR激活突变的局部晚期或转移性NSCLC患者的一线治疗,死亡或疾病进展风险显著降低了41%,中位无进展生存期为14.7个月,总缓解率为75%,中位缓解持续时间为14.8个月[17]。
通常在服用一代或二代EGFR抑制剂后9.2~14.7个月时,患者会出现不同程度的耐药性。在这些耐药突变中,约有50%~70%发生在EGFR上门控位置(Gatekeeper)T790M。该突变可引起EGFR空间构像改变,增加EGFR对ATP的亲和力,从而削弱抑制剂与EGFR的结合能力。二代EGFR抑制剂阿法替尼虽然在体外对EGFR-T790M突变有抑制活性,但在临床应用中仍然未能克服T790M突变产生的耐药性。且一代和二代EGFR抑制剂都很难排除对野生型EGFR的抑制,从而导致明显的皮肤毒性(如痤疮样皮疹)。直到第三代抑制剂奥西替尼出现,这种情况才得以解决,如图1所示。奥西替尼(Osimertinib, AZD9219)和奥母替尼(Olmutinib, HM61713)是针对T790M突变的第三代EGFR-TKIs,对EGFR敏感突变和T790M耐药突变有更好的疗效。奥西替尼为单苯胺基嘧啶环化合物,通过与EGFR激酶区ATP结合域的半胱氨基-797残基不可逆共价结合来抑制EGFR活性[18]。将奥西替尼单药用于一线和二线治疗,疗效良好,具有安全性和可耐受性。Janne等对局部进展或转移的EGFR-TKIs耐药患者的治疗效果、不良反应药效量、有效性及安全性进行了评估[19-21]。而对于联合治疗,体外研究结果显示,如果奥西替尼与 MEK阻滞剂司美替尼和曲美替尼联合使用能有效延迟耐药细胞的生长,意味着奥西替尼与其他抗肿瘤药物联用也许可延缓奥西替尼耐药的发生[22-23]。奥母替尼也是针对T790M突变的第三代EGFR-TKIs,在2016年于韩国批准上市,用于既往接受过TKIs治疗的局部晚期或转移性EGFRT790M突变NSCLC患者。然而治疗病人很高概率发生了第三代的C797S突变,其他突变还有:EGFR L718Q突变、L718Q.L844V和C797S突变,HER2和MET扩增等,使第三代EGFR-TKIs出现了耐药抵抗[24]。EGFR 与 VEGFR均可通过激活下游信号通路PI3K/AKT和Ras/Raf/Erk影响细胞生理功能。在EGFR突变的NSCLC中,上调的 EGFR 突变通过不依赖缺氧的机制增加VEGF,进而促进了对EGFR-TKIs耐药的产生[25]。
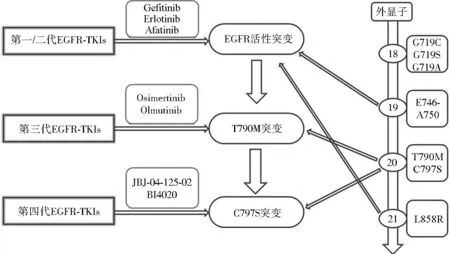
图1 EGFR突变及EGFR-TKIs代表药物
EAI045是第一个针对T790M和C797sEGFR突变的变构非ATP竞争性抑制剂。研究者证明了EAI045能显著提高二聚体缺陷的EGFR突变活性,其与西妥昔单抗联合使用能显著抑制具有L858R/T790M突变的Ba/F3细胞增殖。因为它的残基远离变构结合口袋,C797S突变不会影响EAI045的疗效,但此变构抑制剂单独使用无效,联合使用西妥昔单抗可使EAI045抵制T790M和C797s活性充分提高。西妥昔单抗不是EGFR突变体特异性的单克隆抗体,可能导致靶向WT EGFR相关毒性,所以EAI045未能进入临床试验[26-27]。
2019年,丹娜-法伯癌症研究院研发的JBJ-04-125-02在体外和体内均可抑制细胞增殖和EGFRL858R/T790M/C797S信号传导,而且毒性比EAI045小很多,但JBJ-04-125-02对EGFRDel19/T790M/C797S突变无效。EGFR与其配体结合后,在细胞表面形成二聚体,细胞内自磷酸化后激活下游传导通路。JBJ-04-125-02对EGFR二聚体含量高的肿瘤细胞株抑制能力弱,而西妥昔单抗能够阻断EGFR二聚体的形成,JBJ-04-125-02联合西妥昔单抗增强了抗肿瘤活性。而且,奥希替尼同样可以增强JBJ-04-125-02的抗肿瘤活性,且副作用比西妥昔单抗方案小。2019年底,该研究院对JBJ-04-125-02联合奥希替尼开启了一期临床实验[28]。
2019年10月,我国正大天晴药业集团研发的四代EGFR靶向药TQB3804正式步入I期临床实验。TQB3804不但可以解决奥西替尼的耐药问题,而且能抑制1/2代TKIs耐药后的T790M双重突变,具体如表1所示。
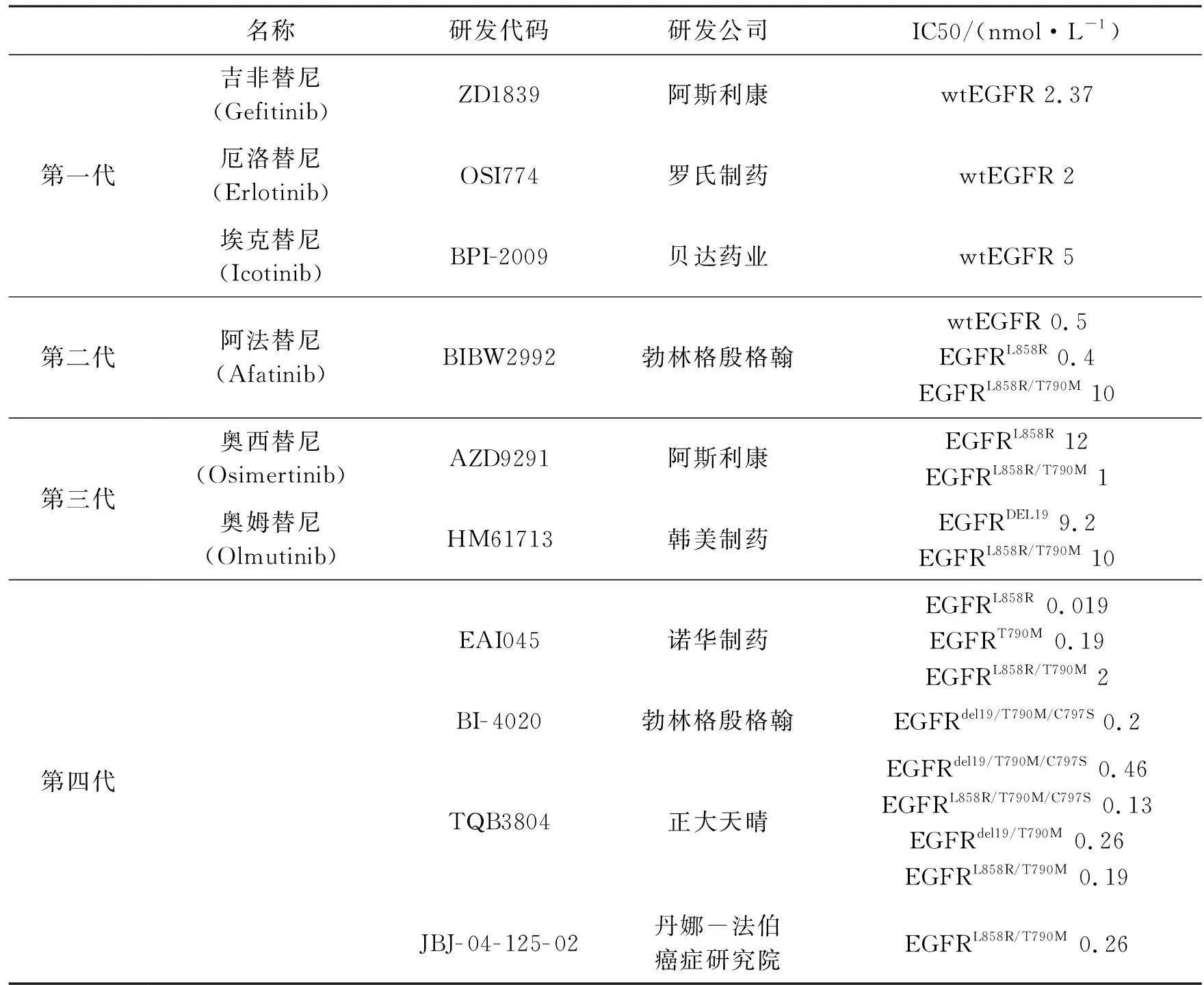
表1 EGFR-TKIs代表药物的基本信息
2019年11月,勃林格殷格翰制药公司的科学家通过高通量筛选和基于结构的药物设计研发了第四代EGFR抑制剂BI-4020。蛋白及细胞实验均显示,其能有效抑制T790M和/或C797S突变的EGFR,并在含顺式三突变del19/T790M/C797S的小鼠异体移植NSCLC肿瘤模型(Human PC-9)上显示很好的药效[29]。
2020年1月,韩国的Bridge Biotherapeutics 公司宣布,美国食品和药物管理局批准了BBT-176作为EGFR靶向的酪氨酸酶抑制剂治疗非小细胞肺癌进入临床实验。
2 EGFR-TKIs与EGFR蛋白的相互作用关系
2.1 第一代EGFR-TKIs
EGFR分子的第790位残基处在非常重要的位置,它位于蛋白结构的连接N和C-lobe的铰链部分,并且处于ATP结合口袋深处, 可以使吉非替尼和厄洛替尼分子与蛋白充分靠近。因此,门控位置 (Gatekeeper)的残基是一个激酶与小分子药物结合的特异性的重要决定因素。吉非替尼和厄洛替尼具有4-苯胺喹唑啉的内核,晶体学结构提示,两种药物对于野生型或突变体激酶结合方式相似,通过与ATP竞争位于激酶区的ATP结合位点,抑制EGFR的酪氨酸激酶活性,如图2和图3所示。发生T790M突变后,蛋氨酸(Met)侧链变长,导致了第一代EGFR-TKIs与ATP口袋结合的位阻性,阻断与药物的最佳相互作用,同时与ATP的亲和力高于EGFR-TKIs,从而使ATP竞争抑制剂失效[30-32]。

图2 吉非替尼的结构及其与EGFR蛋白激酶结构域的键合模式
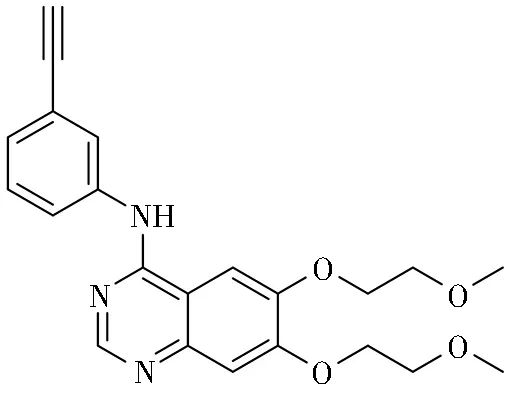
图3 厄洛替尼的结构及其与EGFR蛋白激酶结构域的键合模式
2.2 第二代EGFR-TKIs
阿法替尼的喹唑啉环是重要的结构单元,与ATP竞争细胞内的激酶结构域,从而阻碍下游信号的传导。 喹唑啉母核上的N-1原子与激酶Met793上的NH形成分子间氢键。喹唑啉环C4位上的2-氯-3氟苯胺基与空间上方Val726和Met766的分子间作用力使阿法替尼进一步深入ATP结合位点。喹唑啉母核C6位上侧链的烯丙酰结构对Afatinib发挥抗肿瘤活性作用至关重要,它与EGFR上半胱氨酸残基Cys797的巯基发生迈克尔加成反应,可使激酶失活,不可逆地抑制酪氨酸激酶的活性,如图4所示[33-34]。研发人员通过结构鉴定进一步证明了共价键的存在,并且发现阿法替尼通过与HER2的Cys805以及HER4的Cys803相互作用而抑制酶的活性[35]。

图4 阿法替尼的结构及其与EGFR蛋白激酶结构域的键合模式
2.3 第三代EGFR-TKIs
奥西替尼和奥母替尼均具有一个“U”型结构,有利于与表皮因子受体目标结合。第三代EGFR-TKIs有丙烯酰胺的结构单元,它可以作为受体与在表皮因子受体ATP结合域内的Cys797的活性巯基发生迈克尔加成反应形成共价键,而带有C797S突变的EGFR不能形成共价键。因为在生理条件下,丝氨酸的无活性巯基不能形成共价键,提供一个关键机制的阻力[9]。在晶体结构中,奥西替尼结合在P-loop和Pro794、Cys797之间的蛋白质骨架的ATP结合口袋外边缘。有两个氢键帮助锚定配体,即奥西替尼的N4氢原子与Met793的N原子;羰基氧原子与Cys797的N原子结合形成两个氢键帮助固定配体。奥西替尼的取代苯环,与下面的蛋白质骨架发生范德华相互作用,与上面的非极性残基广泛作用,完成了夹心效果,Leu718与奥西替尼的苯环接触,Ala743与嘧啶环接触,Val726和Phe723与吲哚环接触,Leu718-Gly719的蛋白质骨架也与吲哚环的边缘形成范德华相互作用力,如图5所示[36]。

图5 奥西替尼的结构及其与EGFR蛋白激酶结构域的键合模式
同为第三代EGFR-TKIS的还有艾氟替尼(Alflutinib, AST2818),它是上海爱利斯特制药有限公司开发的一种基于三氟乙氧基吡啶的不可逆EGFR-TKIs,对EGFR耐药突变(如G719X、19号外显子缺失、L858R、L861Q和T790M)具有较高的选择性,对野生型EGFR无抑制作用。
2.4 第四代EGFR-TKIs
奥西替尼虽然解决了T790M突变的问题,但临床上已经观察到,进行奥西替尼二线治疗的EGFRT790M阳性NSCLC患者,服药10个月后出现耐药现象,其中20%~40%为C797S突变(包含del19/T790M/C797S或L858R/T790M/C797S的顺式或反式三突变)。因此,开发下一代EGFR抑制剂需要满足更多临床需求:一是对del19突变(最常见的突变形式)的EGFR具有高活性;二是能解决耐药突变T790M和/或C797S;三是对野生型EGFR无作用;四是在整个人类激酶组显示高选择性。EAI045与EGFR作用呈“Y”形或“三叶螺旋桨”形,从EGFRT790M突变激酶的共晶结构可以看出,抑制剂EAI001与经典的ATP结合口袋附近的变构口袋结合,部分通过由内向外形成αC-螺旋结构,结合后作为一个“三叶螺旋桨”或“Y”形配置。氨噻唑片段延伸至T790M守门人蛋氨酸与Lys745残基活性位点之间,视为T790M突变体的选择性;苯基延伸到ATP结合口袋后面的疏水空隙,与Leu-777和Phe-856残基相互作用,而第三个区域,1-氧异吲哚基沿αC-螺旋结构向溶液暴露区延伸。另外,EAI001氨基组在Asp-Phe-Gly(DFG)序列与Asp-855之间形成氢键。进一步药物化学优化使苯基组产生了更大的影响力,也就是EAI045对野生型EGFR保持千倍的选择性。然而EAI045的结合位点是EGFR激酶在不活跃的构象时由αC-螺旋运动产生的。两个亚基的不对称二聚体构成的异构结合位点不可接近,因此单独使用EAI045的疗效是有限的。当与西妥昔单抗联合使用时,EAI045能成功到达每一种EGGFR激酶结构域[37-38],EAI045的噻唑、异吲哚啉-1-酮和对氟苯酚的芳香环,与EGFR激酶的多个疏水基团进行交互作用。具体来说,噻唑基团与Met790侧链相互作用,对氟苯酚的苯基与Phe856侧链相互作用,异吲哚啉-1-酮基团与Lle759、Leu747、Leu788、Leu777和Met766侧链发生广泛的疏水性相互作用,如图6所示。极性在EAI045与EGFR激酶的相互作用中起到了重要作用。化合物的酰胺氮原子与EGFR的Asp855主链羧基形成氢键,异吲哚啉-1-酮的羰基氧与Lys745的侧链ε-胺基形成另一个氢键[39-40]。
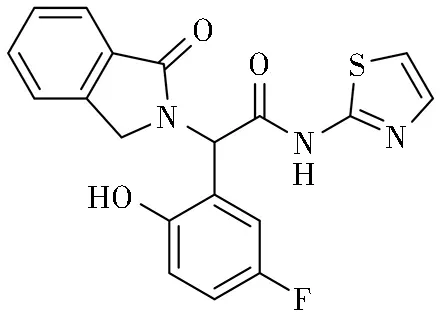
图6 EAI045的结构及其与EGFR蛋白激酶结构域的键合模式
另外,吡啶并[3,4-d]嘧啶类化合物可以作为新的EGFR-TKIs[41-42]。吡啶并[3,4-d]的7位氮原子和6位的氨基吡啶氢原子,在铰链区域与Met793形成了两个氢键,支架2位的苯胺氢原子与看门人残基Met790形成氢键,4位的氨基吡啶或氨基哌啶占据ATP核糖袋,并从Asp-Phe-Gly序列与Phe856和Asp产生范德华相互作用。位于吡啶并[3,4-d]嘧啶2位的苯胺插入背侧疏水袋(BHP)产生疏水作用,这个疏水作用是必要的。4位的氨基哌啶上含有羟基,能使突变Ser797的氢键作用增强,能够被设计成针对Ser797突变的小分子抑制剂。
JBJ-04-125-02的结构式如图7(a)所示,它能够与EGFR的变构袋结合,这种变构袋是由失活激酶构象中轮状核c-螺旋向外位移产生的,如图7(b)所示。噻唑酰胺、苯环和异吲哚啉酮的结合模式与之前观察到的EAI001相似[40]。此外,在DFG基序中,羟基与Phe856的羰基形成氢键。4-哌嗪基苯基沿着环-螺旋延伸到溶剂暴露外表面,其苯环在激酶p-环上与Phe723发生π-π堆积。有趣的是,该化合物的结合诱导了激酶激活环的新构象,激活环中哌嗪和Glu865之间的氢键稳定了该构象,如图7(c)所示。此外,Glu749定位在哌嗪基团的氢键上。预计与EAI045相比,重新配置的活化环和与哌嗪基团形成的氢键有助于增强该化合物的效力[28]。
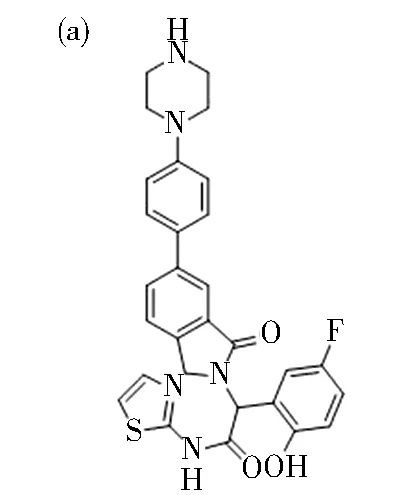
图7 (a) JBJ-04-125-02化学结构式; (b)与JBJ-04-125-02和AMP-PNP结合的 EGFRT790M/V948R晶体结构; (c)变构结合口袋
3 展 望
随着医疗技术的不断发展,人们对肿瘤的致病机制进一步了解,靶向治疗成为研究热点。不断发展的靶向药物能够抑制酪氨酸激酶在细胞内的信号传导,在肿瘤发生侵袭时发挥重要作用,针对EGFR-TKIs开发的一系列分子靶向抑制剂将会为肿瘤患者带来希望。另外,随着血液样本中ctDNA检测技术的快速发展,癌症个体化深度测序分析方法(Cancer personalized profiling by deep sequencing, CAPP-Seq)已经能够准确测定非小细胞肺癌突变基因的分子分型,指导EGFR-TKIs靶向药物的选用。未来基因检测技术将适用于多种类型的恶性肿瘤并广泛应用于临床,相信会有更多疗效精准的靶向药物问世,促进个体化癌症治疗的发展,改善人类的健康状态。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
天然产物研究与开发(2018年1期)2018-02-02
中成药(2018年1期)2018-02-02
中国塑料(2016年7期)2016-04-16
中学化学(2015年12期)2016-01-19
中国医药生物技术(2015年4期)2015-12-26
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年1期)2014-02-28
吉林大学学报(医学版)(2014年2期)2014-02-27
