
4-F-α-PVP 类似物4-F-3-Methyl-α-PVP 盐酸盐的结构解析与表征
2023-07-06 04:31王昊阳吴剑张倩闵新峰刘秀艳郭寅龙
法医学杂志 2023年2期
王昊阳,吴剑,张倩,闵新峰,刘秀艳,郭寅龙
中国科学院上海有机化学研究所,上海 200032
目前,新精神活性物质(new psychoactive substance,NPS)在世界范围内不断涌现,已成为国际禁毒领域公认的难题。NPS 又称策划药以及毒品类似物,一部分是不法分子为逃避打击,通过对列管精神药物或毒品的结构进行修饰而得到的类似结构化合物,另一部分则是设计或筛选出来的,是精神活性物质向毒品转变的过渡形态[1]。NPS 生产简单、违法成本较低、交易形式隐蔽,在欧美等国家呈快速蔓延之势。NPS 的种类也在不断变化,因滥用危害严重,越来越多的NPS 被列管为毒品[2]。
合成卡西酮类的结构类似于苯丙胺,主要是对苯丙胺类化合物分子骨架上的芳环、α 碳和胺基进行了修饰,可以产生大量结构类似的化合物。这些修饰后的卡西酮类似物被认为可能具有更大的药物依赖性和毒性风险[3]。卡西酮类似物的滥用与欣快、好斗、暴力行为、心动过速、癫痫发作、幻觉、兴奋、谵妄甚至死亡有关[4-5]。1-(4-氟苯基)-2-(N-吡咯烷基)-1-戊酮[1-(4-fluorophenyl)-2-(1-pyrrolidinyl)pentan-1-one,4-F-α-PVP;CAS 号为850352-62-4]、1-(4-甲氧基苯基)-2-(N-吡咯烷基)-1-戊酮[1-(4-methoxyphenyl)-2-(1-pyrrolidinyl)pentan-1-one,4-MeO-α-PVP;CAS号为14979-97-6]和1-苯基-2-(N-吡咯烷基)-1-戊酮[1-phenyl-2-(1-pyrrolidinyl)pentan-1-one,α-PVP;CAS 号为14530-33-7]均属于2015 年10 月1 日起实施的《非药用类麻醉药品和精神药品列管办法》[6]中的管制品种(化学结构见图1),2021 年7 月9 日相关标准[7]发布,规定了法庭科学领域疑似毒品样品中1-苯基-2-(N-吡咯烷基)-1-丁酮[1-phenyl-2-(1-pyrrolidinyl)butan-1-one,α-PBP]、α-PVP 和4-F-α-PVP 的液相色谱-质谱定性检验方法和液相色谱定量检验方法。本研究在没有对照品的情况下,借鉴文献[8-12]报道,对新近缴获的检材进行综合谱学表征与分析,拟为法庭科学实验室在鉴别和分析该化合物以及类似化合物时提供参考。
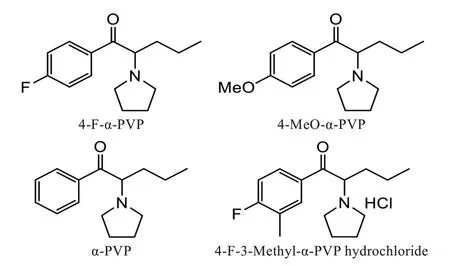
图1 4-F-α-PVP 及其类似物的化学结构Fig.1 Chemical structures of 4-F-α-PVP and its analogs
1 材料与方法
1.1 样本
未知化合物由公安机关在缉毒工作中缴获,外观为白色晶体,可研磨成白色粉末。
1.2 主要仪器与试剂
5973N 质谱仪(美国Agilent公司);GCMS-QP2010 Ultra 气相色谱质谱联用仪(日本岛津公司);Q Exactive HF 静电场轨道阱超高分辨质谱仪(美国Thermo Fisher Scientific 公司),配电喷雾离子源(electrospray ionization,ESI)和VanquishTM超高效液相色谱(ultra-high performance liquid chromatography,UPLC);AVANCE NEO 500 MHz核磁共振波谱仪(德国Bruker公司);DionexTMAquionTM离子色谱系统(美国Thermo Fisher Scientific 公司);Nicolet iZ10 红外光谱仪(美国Thermo Fisher Scientific 公司);Milli-Q Advantage A10 超纯水系统(美国Millipore 公司)。
甲醇、乙腈、甲酸(色谱纯,德国Merck 公司),氘代氯仿(CDCl3,99.8%;美国剑桥同位素实验室)。
1.3 仪器条件
1.3.1 直接进样电子电离-质谱(electron ionizationmass spectrometry,EI-MS)分析条件
直接进样杆采用程序升温,正离子模式,离子源的电子能量为70 eV,质量扫描范围为m/z35~500。
1.3.2 GC-MS 分析条件
J&W DB-5ms 石英毛细管柱(30 m×0.25 mm,0.25 μm;美国Agilent 公司)。升温程序:140 ℃保持3 min,20 ℃/min升至300 ℃,保持13 min。载气为氦气,流速1 mL/min,分流比为20∶1,溶剂延迟3 min,进样口温度280 ℃,EI 源电子能量70 eV,离子源温度230 ℃,传输线温度250 ℃,质量扫描范围为m/z35~500。
1.3.3 电喷雾离子化-高分辨质谱(electrospray ionizationhigh resolution mass spectrometry,ESI-HRMS)分析条件
样品溶液直接进样分析,流动相A 为0.1%甲酸水溶液,流动相B 为甲醇,正离子全扫描及串联质谱扫描;喷雾电压3.5 kV,喷雾温度350 ℃,雾化气(N2)35 Arb,辅助气(N2)15 Arb;一级质谱扫描范围为m/z100~1 000,二级质谱碰撞能量10~40 eV。
1.3.4 超高效液相色谱-高分辨串联质谱(ultra-high performance liquid chromatography-high resolution tandem mass spectrometry,UPLC-HRMS/MS)分析条件
ACQUITY UPLC BEH C18色谱柱(2.1 mm×100 mm,1.7 μm;美国Waters公司),柱温40 ℃,流动相A为0.1%甲酸水溶液,流动相B 为乙腈。梯度洗脱程序:初始为5%B,0~8.0 min 5%B~90%B,8.0~9.5 min 90%B,9.5~9.6 min 90%B~5%B,9.6~12 min 5%B。流速为0.4 mL/min,进样量为1 μL。采用ESI 离子化方式,喷雾电压3.5 kV,喷雾温度350 ℃,雾化气(N2)35 Arb,辅助气(N2)15 Arb;正离子串联质谱扫描,串联质谱碰撞能量40 eV。
1.3.5 核磁共振(nuclear magnetic resonance,NMR)分析条件
对样品分别进行核磁共振氢谱(1H-nuclear magnetic resonance,1H-NMR;500 MHz)、核磁共振碳谱(13C-nuclear magnetic resonance,13C-NMR;126 MHz)、氢-碳异核单量子相关谱(1H-13C heteronuclear single quantum correlation spectroscopy,1H-13C HSQC)和异核多键碳氢相关谱(1H-13C heteronuclear multiple bond correlation spectroscopy,1H-13C HMBC)分析。1H的化学位移参考氘代氯仿中残留的氯仿峰定为δ=7.26,13C 的化学位移参考氘代氯仿的峰定为δ=77.16。
1.3.6 离子色谱分析条件
分析柱DionexTMIonPacTMAS14A(4 mm×250 mm)和保护柱DionexTMIonPacTMAG14(4 mm×50 mm;美国Thermo Fisher Scientific 公司);柱温30 ℃,淋洗液为0.75 mmol/L NaHCO3和6.0 mmol/L Na2CO3,检测方式为电导检测器,流速1.0 mL/min,进样量25 μL。
1.3.7 傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)分析条件
样品采用溴化钾压片法进行FTIR 分析,采集范围为4 000~400 cm-1,波长分辨率为4 cm-1,采样32次。
1.4 样品处理
取微量样品,首先进行直接进样EI-MS 分析。再以甲醇为溶剂,将样品配制成质量浓度约1 μg/mL 的溶液,供GC-MS 分析。用初始流动相将样品配制成质量浓度约1 μg/mL 的溶液,供ESI-HRMS 与UPLCHRMS/MS分析。取适量样品溶解于1 mL氘代氯仿中,供NMR 分析。精确称取5 mg 样品溶于10 mL 容量瓶中,加入去离子水溶解、稀释、定容,供离子色谱分析。
2 结果与讨论
2.1 直接进样EI-MS 和GC-MS 分析
该样品直接进样EI-MS 分析的质谱图(图2A)中主要离子见表1,EI-MS 谱图中的基峰为m/z126。样品GC-MS 分析的总离子流色谱图见图2B,主成分保留时间为7.95 min。将EI-MS谱图(图2A)与美国国家标准与技术研究院(National Institute of Standards and Technology,NIST)质谱数据库(https://doi.org/10.18434/T4D303)进行比对和检索,尽管未检索到完全相匹配的结果,但是检索得到的一系列可能的类似化合物结构,给该未知化合物的结构解析提供了重要线索。

表1 未知化合物EI-MS 谱图中主要的离子Tab.1 Major ions of the unknown compound in EI-MS spectrum
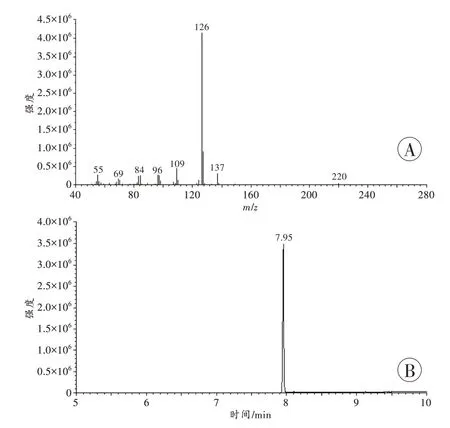
图2 未知化合物的EI-MS谱图和GC-MS总离子流色谱图Fig.2 EI-MS spectrum and GC-MS total ion chromatogram of the unknown compound
NIST 质谱数据库检索推荐的可能结构为4-Methyl-α-PVP,该化合物EI-MS 谱图中也含有m/z126 的基峰[12],是分子离子发生α 裂解反应丢失中性4-甲基苯甲酰基自由基,形成的1-丁烯基吡咯1-亚胺正离子m/z126,对应的分子式为C8H16N+,而且4-F-α-PVP 的EI-MS 谱图中的基峰也是m/z126。进一步分析发现:4-Methyl-α-PVP 在EI-MS中发生α 裂解反应得到丰度较低的4-甲基苯甲酰基正离子m/z119,对应的可能是未知化合物EI-MS 谱图中的碎片离子m/z137,结合对4-F-α-PVP EI-MS 谱图中类似裂解过程产生的4-氟苯甲酰基正离子m/z123 进行分析,推测该未知化合物极有可能为苯环上多了1 个甲基的4-F-α-PVP 结构类似物。随后对该样品进行了ESI-HRMS、UPLC-HRMS/MS、NMR、FTIR 和离子色谱分析,来解析其准确的结构。
2.2 ESI-HRMS 和UPLC-HRMS/MS 分析
在正离子化模式下,样品的ESI-HRMS 谱图(图3A)主要显示m/z264 的离子,预测其分子式为C16H23FNO+,相对误差仅为0.38×10-6。这一结果进一步支持了该化合物是4-F-α-PVP 的苯环上多了1 个甲基的推测。m/z264 离子在UPLC-HRMS/MS 谱图(图3B)中主要产物离子的分子式分析结果(表2)也支持这一推测。样品的UPLC-HRMS/MS 总离子流色谱图见图3C,主成分的保留时间为4.72 min。
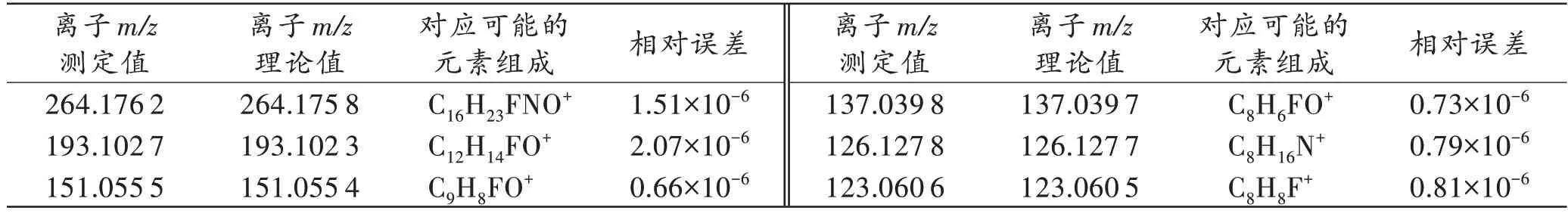
表2 m/z 264 离子在UPLC-HRMS/MS 谱图中的主要产物离子Tab.2 Major product ions of the ion at m/z 264 in UPLC-HRMS/MS spectrum
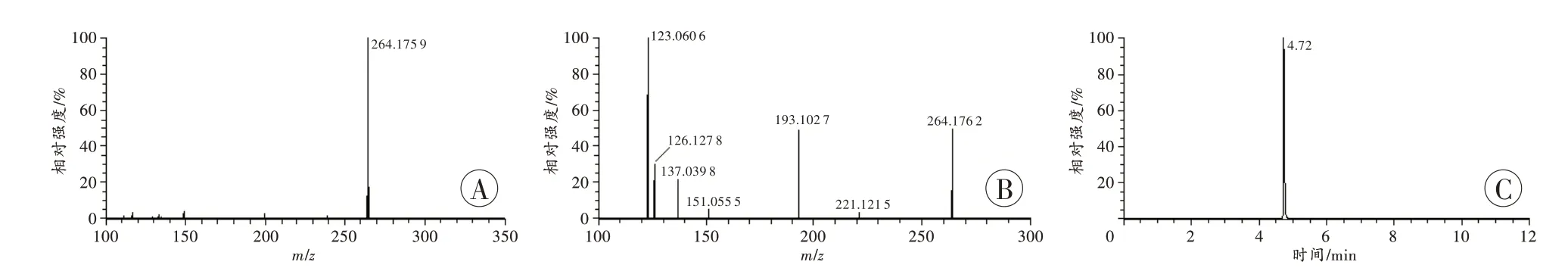
图3 未知化合物的ESI-HRMS 和UPLC-HRMS/MS 谱图Fig.3 ESI-HRMS and UPLC-HRMS/MS spectra of the unknown compound
2.3 NMR 分析
样品的EI-MS 谱图(图2A)和ESI-HRMS/MS 谱图(图3B)都表明了该未知化合物可能为苯环上含甲基的4-F-α-PVP 结构类似物,苯环上甲基和氟原子的具体位置需要进一步分析确认。
样品的1H-NMR、13C-NMR、1H-13C HMBC 和1H-13C HSQC 谱图分别见图4。表3 总结了样品1H-NMR谱图(图4A)和13C-NMR 谱图(图4B)中化学位移的信息。
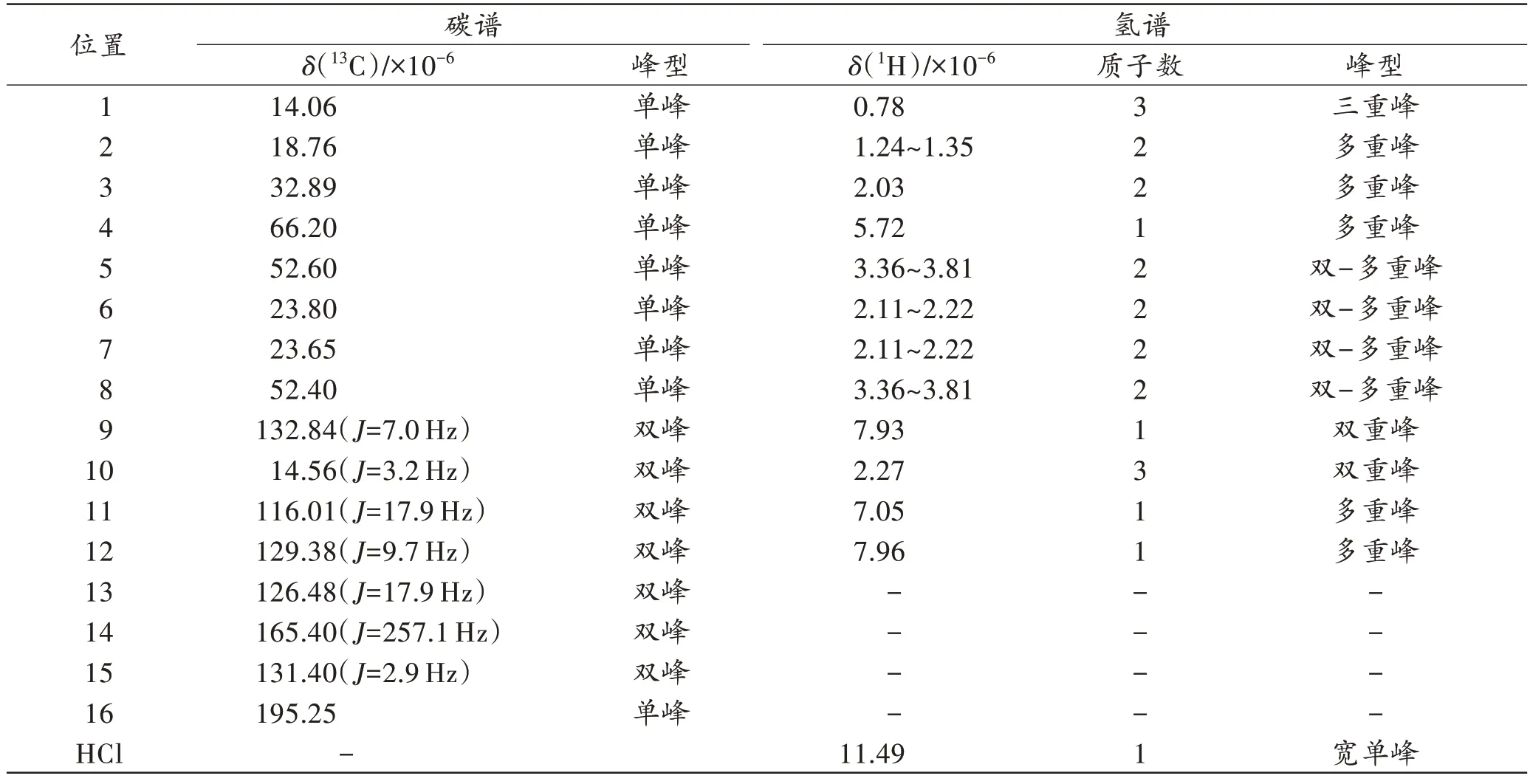
表3 未知化合物1H-NMR 和13C-NMR 的化学位移值Tab.3 1H-NMR and 13C-NMR chemical shift of the unknown compound
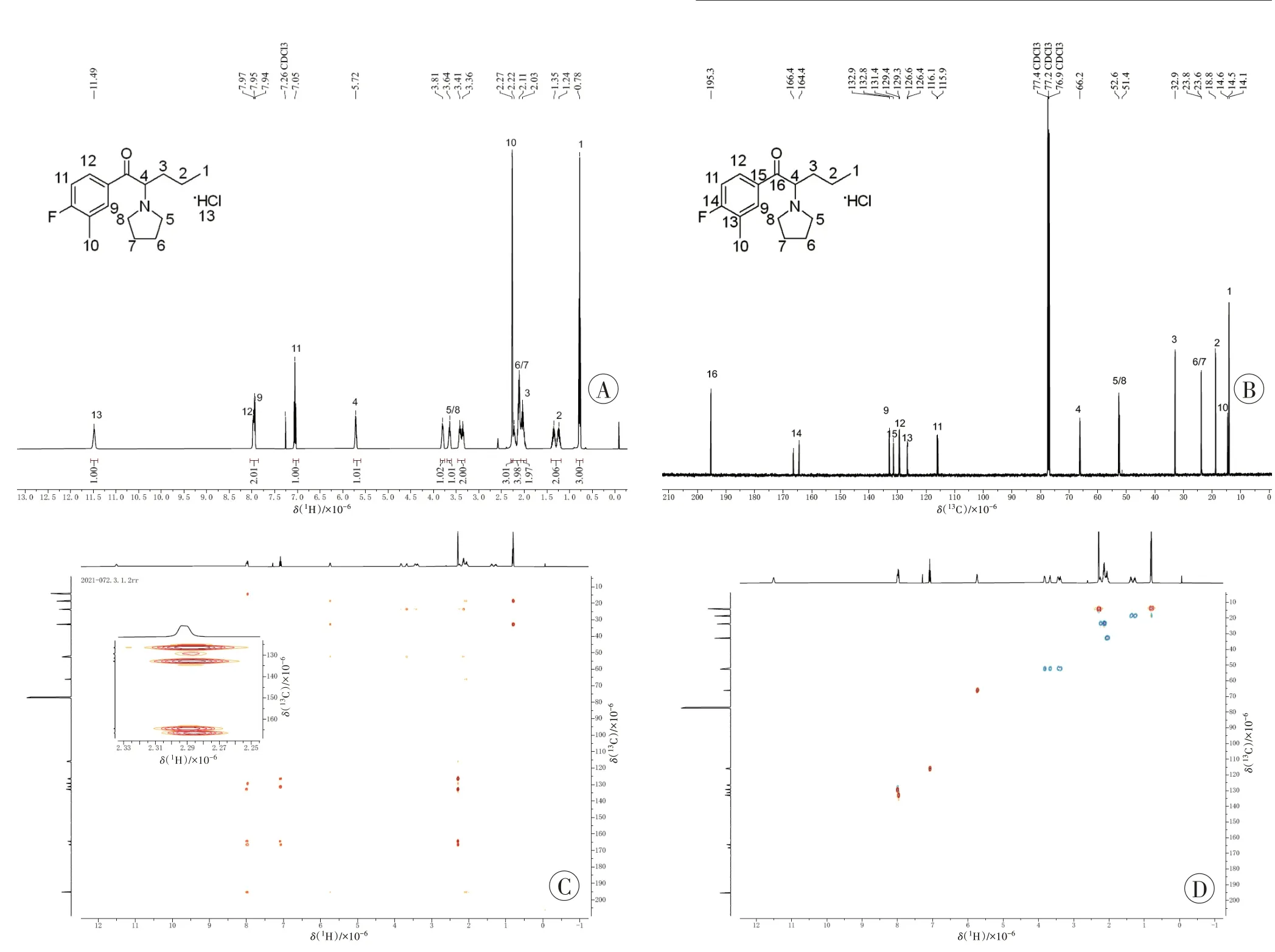
图4 未知化合物的1H-NMR、13C-NMR、1H-13C HMBC、1H-13C HSQC 谱图Fig.4 1H-NMR,13C-NMR,1H-13C HMBC,1H-13C HSQC spectra of the unknown compound
甲基与氟原子的位置经NMR 分析得到了确认。在1H-13C HMBC 谱图(图4C)中,C-16(δ=195.25)与H-9(δ=7.93)和H-12(δ=7.96)的相关信号表明甲基与氟原子不在C-9 和C-12 位。在13C-NMR 谱图(图4B)中,芳香区氟对碳的耦合常数(J)和碳的种类分析如下:C-14(δ=165.40,J=257.4 Hz,符合13C-19F 自旋耦合常数,季碳),C-13(δ=126.48,J=17.9 Hz,季碳),C-11(δ=116.01,J=17.9 Hz,叔碳),C-9(δ=132.84,J=7.0 Hz,叔碳),C-12(δ=129.38,J=9.7 Hz,叔碳),C-15(δ=131.40,J=2.9 Hz,季碳),表明氟原子和羰基处于对位。同时,在1H-13C HMBC 谱图(图4C)中,H(Me)-10(δ=2.27)与C-14(δ=165.40,J=257.4 Hz,季碳)、C-13(δ=126.48,J=17.9 Hz,季碳)和C-9(δ=132.84,J=7.0 Hz,叔碳)的相关信号(1H-13C HMBC 谱图中放大的部分)表明甲基处于氟原子邻位。1H-13C HSQC 谱图(图4D)中,H-11(δ=7.05)与C-11(δ=116.01,J=17.9 Hz)的相关信号表明甲基和氟原子的位置不在C-11 位。
基于以上数据可初步推断未知化合物主体结构为1-(4-氟-3 甲基苯基)-2-(N-吡咯烷基)-1-戊酮[1-(4-fluoro-3-methyl phenyl)-2-(1-pyrrolidinyl)pentan-1-one,4-F-3-Methyl-α-PVP],由于1H-NMR分析发现实际氢的个数比4-F-3-Methyl-α-PVP 中性分子多1个(在δ=11.49,宽单峰),可能对应的是1个活泼氢,因此推断该化合物以盐形式存在。随后使用离子色谱法进行分析,表明该化合物含氯离子(含量11.14%~11.16%),这一结果进一步支持了该化合物是一种盐酸盐。
2.4 FTIR 分析
样品的FTIR 分析结果见图5。从FTIR 谱图可以看出:1 682 cm-1处有强尖峰,说明化合物中含有羰基;3 033、3 015 cm-1处为苯环的C-H 吸收峰,1 601~1 450 cm-1处为苯环的振动吸收峰;2 960~2 870 cm-1处为亚甲基和甲基的振动吸收峰;1 339 cm-1处出现C-N-C 吸收峰,3 346 cm-1处为N-H 振动峰,说明该未知化合物中有含氮结构,且2 624 cm-1处为铵盐吸收峰。
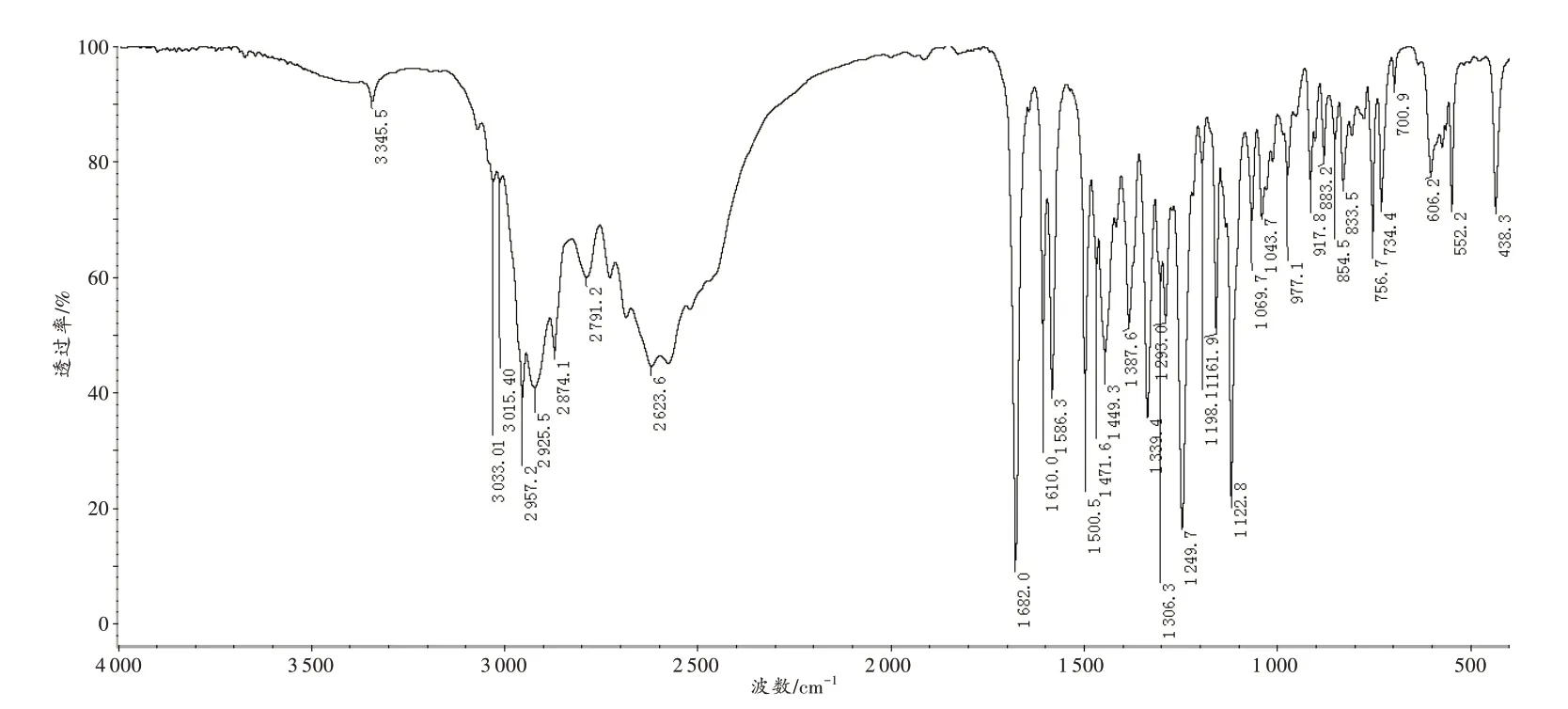
图5 未知化合物的FTIR 谱图Fig.5 FTIR spectrum of the unknown compound
综合样品的质谱分析(图2~3,表1~2)、核磁数据信息归属(图4,表3)、FTIR 分析(图5)和离子色谱分析结果,最终确定该未知化合物为4-F-3-Methylα-PVP 盐酸盐。
2.5 化合物质谱裂解规律分析和结构确证
4-F-3-Methyl-α-PVP 在EI-MS 模式下可能的裂解途径见图6。EI-MS 谱图中的基峰m/z126 可能是4-F-3-Methyl-α-PVP 在EI-MS 模式下发生α 裂解丢失中性4-氟-3-甲基苯甲酰基自由基,形成的1-丁烯基吡咯1-亚胺正离子m/z126,对应的分子式为C8H16N+;或生成正电荷在4-氟-3-甲基苯甲酰基的产物离子m/z137,对应的分子式为C8H6FO+,该离子还可以进一步脱CO 生成产物离子m/z109,对应的分子式为C7H6F+。m/z126 碎片离子同样存在于4-MeOα-PVP、α-PVP 和4-F-α-PVP 的EI-MS 谱图中[13],该离子还可以进一步生成m/z96、m/z84 和m/z55 产物离子。4-F-3-Methyl-α-PVP 在EI-MS 模式下还可以发生α 裂解丢失中性丙基自由基,生成m/z220 的碎片离子。
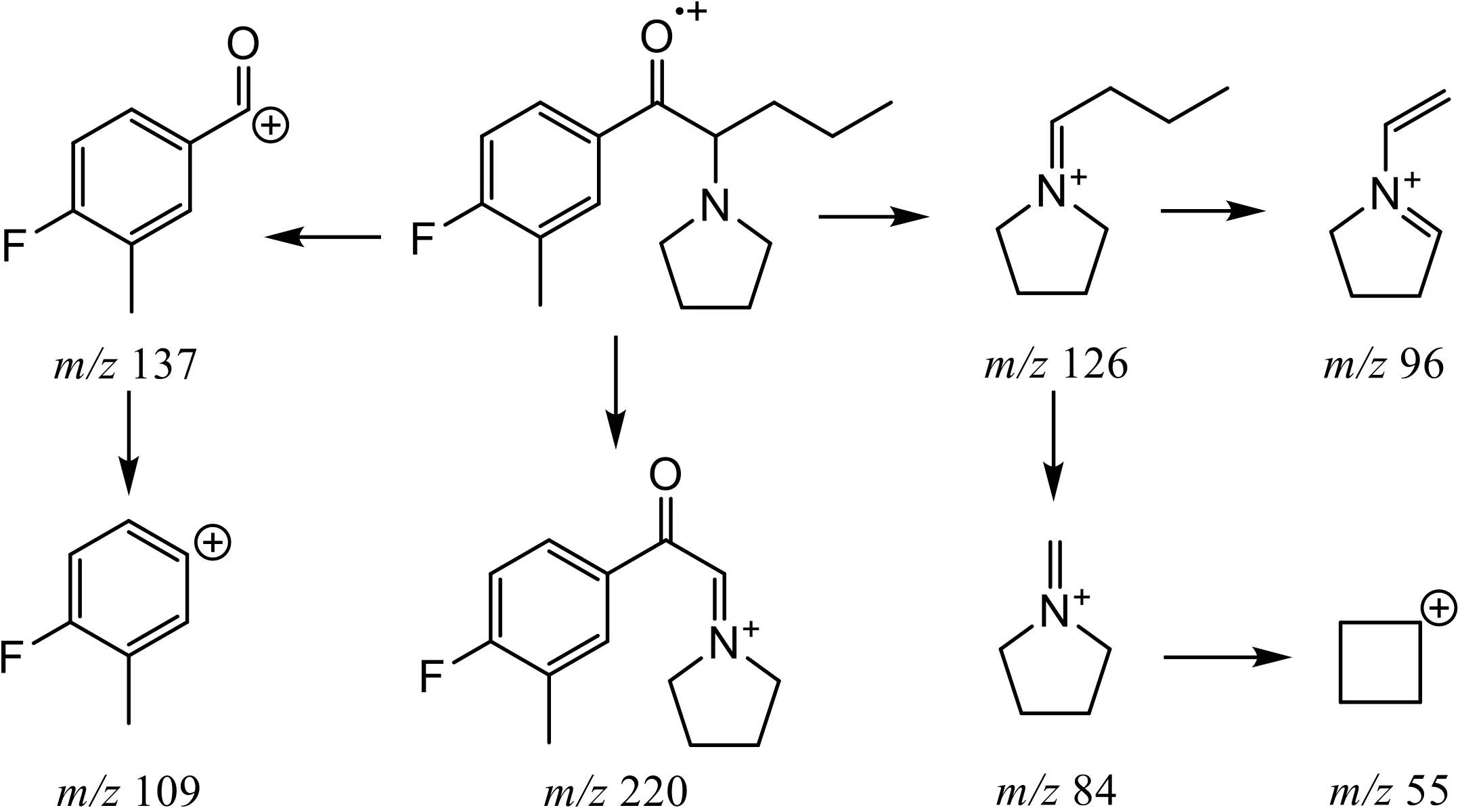
图6 EI-MS 模式下4-F-3-Methyl-α-PVP可能的裂解途径Fig.6 Proposed fragmentation pathways of 4-F-3-Methyl-α-PVP in EI-MS mode
4-F-3-Methyl-α-PVP 在UPLC-HRMS/MS 模式下可能的裂解途径见图7。m/z264离子主要发生丢失中性四氢吡咯的裂解反应,生成产物离子m/z193,这是4-F-3-Methyl-α-PVP 在UPLC-HRMS/MS 模式下特有的产物离子[12-13],在4-F-3-Methyl-α-PVP 的EIMS 谱图中未发现,与4-F-α-PVP 在UPLC-HRMS/MS模式下的产物离子m/z179 对应[13]。m/z193 离子进一步丢失中性丙烯分子得到产物离子m/z151,随后其再脱CO 生成相对丰度最高的产物离子m/z123。与4-F-α-PVP 在UPLC-HRMS/MS 模式下的裂解过程类似,产物离子m/z137 进一步脱CO 生成产物离子m/z109[13]。此外,4-F-3-Methyl-α-PVP 在UPLCHRMS/MS 模式下也可以生成4-氟-3-甲基苯酰基正离子m/z137和1-丁烯基吡咯1-亚胺正离子m/z126,这与4-F-3-Methyl-α-PVP 在EI-MS 中的碎片离子相同。

图7 UPLC-HRMS/MS 模式下4-F-3-Methyl-α-PVP可能的裂解途径Fig.7 Proposed fragmentation pathways of 4-F-3-Methyl-α-PVP in UPLC-HRMS/MS mode
3 结论
新出现的NPS 对公共健康存在巨大的威胁,也对法庭科学实验室的鉴定能力提出了挑战。本研究以4-F-3-Methyl-α-PVP盐酸盐为例,详细介绍了在没有对照品参考的情况下如何对未知化合物进行结构解析。本研究通过综合分析EI-MS、GC-MS、ESI-HRMS、UPLC-HRMS/MS、NMR、离子色谱法和FTIR 分析提供的质谱、色谱和波谱信息,最终确定了缴获的未知化合物为4-F-3-Methyl-α-PVP 的盐酸盐。本研究还对该化合物在EI-MS 和UPLC-HRMS/MS 分析条件下生成的碎片离子进行了裂解途径的推导,这些信息将有助于其他法庭科学实验室对4-F-3-Methyl-α-PVP盐酸盐及其类似物进行结构解析和检测。
猜你喜欢
——碳正离子的产生及稳定性比较
高中数理化(2022年20期)2022-11-17
肝博士(2022年3期)2022-06-30
数理化解题研究·高中版(2022年10期)2022-05-30
食品安全导刊(2021年20期)2021-08-30
天津化工(2020年5期)2020-10-15
当代化工研究(2016年5期)2016-03-20
医学研究杂志(2015年6期)2015-07-01
特产研究(2014年4期)2014-04-10
化学教学(2014年1期)2014-02-20
