
牙龈卟啉单胞菌与心血管疾病致病机制的相关进展
2023-06-21 02:47胡颖文马晓岚牛海涛黄跃
暨南大学学报(自然科学与医学版) 2023年2期
胡颖文,马晓岚,牛海涛,黄跃,3*
(1.暨南大学 口腔医学院,广东 广州 510632;2.暨南大学 附属口腔医院,广东 佛山 528300;3.广州市无菌动物与微生态转化重点实验室,广东 广州 510632;4.暨南大学 基础医学与公共卫生学院,广东 广州 510632;5.暨南大学 实验动物管理中心,广东 广州 510632)
慢性牙周病影响着全球近50%的人口,许多研究发现它与全身系统疾病有着密切的关联,特别是与约占全球人类所有死亡的1/3的心血管系统疾病有着显著的相关性。而牙龈卟啉单胞菌(Porphyromonasgingivali)是定植于牙周袋中的绝对厌氧菌,是牙周炎的常见致病菌,与心血管疾病(cardiovascular disease,CVD)的发生发展密切相关。Porphyromonasgingivalis及其释放的毒性产物可通过引发机体一系列的免疫炎症反应,使得炎症介质进入血液循环,加速动脉粥样硬化的进展、诱导免疫反应加重高血压、抑制梗死心肌的愈合等,从而影响CVD的发生和发展[1-2]。随着研究的深入,本综述详细地总结了Porphyromonasgingivalis与CVD的相关性,揭示了不同致病因子、途径、机制之间导致的不同结果,为今后Porphyromonasgingivalis与CVD相关性研究提供新的研究视角。
1 牙龈卟啉单胞菌的致病因子
Porphyromonasgingivalis是一种机会致病菌,其通过逃避宿主免疫监视来干扰宿主防御机制,通过致病因子入侵宿主细胞导致致病。其致病因子包括外膜囊泡(outer membrane vesicles,OMVs)、脂多糖(lipopolysaccharide,LPS)、牙龈蛋白酶、菌毛、荚膜等[3]。它们单独或一起发挥着作用,加重CVD的发生[4]。
1.1 外膜囊泡
OMVs直径在 50 至 250 nm 之间,平均约为 50 nm[5],其内部含有LPS、外膜蛋白、磷脂、热休克蛋白和DNA[6]。OMVs高度集中大量的毒性因子,避免了蛋白水解酶的降解和破坏,增强了Porphyromonasgingivalis的毒性。Farrugia等[7-8]研究表明,OMVs可通过牙龈蛋白酶水解切割人血管内皮细胞(endothelial cells,ECs),血管通透性增加,导致组织渗出液增加,组织水肿 。OMVs通过抑制一氧化氮,血管收缩增加,内皮细胞凋亡增多,促进AS的发生[9]。OMVs还参与了细菌黏附,生物膜形成、侵袭并损伤宿主细胞和从而影响宿主免疫应答的调节[4,10-11]。
1.2 脂多糖
LPS是细菌外膜的一个相对分子量约为10 kD的较大的分子,是细菌外膜的重要组成部分,对牙周组织具有很强的毒性作用,是Porphyromonasgingivalis的主要毒力因子。LPS可以在Porphyromonasgingivalis存活期间游离成膜泡释放出来,也可以在Porphyromonasgingivalis死亡裂解后释放出来,透过组织的上皮屏障,侵入并破坏深层组织。Porphyromonasgingivalis-LPS及Porphyromonasgingivalis菌毛,能够诱导不同Toll样受体(Toll-like receptors,TLR),发生炎症和免疫反应,并且识别TLR2并结合蛋白LBP对机体CD14受体等信号通路诱导成骨细胞产生肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)等炎症因子诱导牙槽骨溶解破坏[12-14]。
1.3 牙龈蛋白酶
牙龈蛋白酶可以直接损伤牙周组织,并且辅助Porphyromonasgingivalis逃逸宿主免疫系统使其长时间持续存在并破坏牙周组织。牙龈蛋白酶诱导基质金属蛋白酶表达量过大,基质金属蛋白酶组织抑制因子过小,从而致使牙周组织细胞间隙扩大,通透性增强,开辟通道使细菌及其毒性产物入侵,破坏结缔组织并引起牙周组织变性、降解,加深牙周袋和牙槽骨的吸收[15-17]。
1.4 菌毛
Porphyromonasgingivalis菌毛是Porphyromonasgingivalis的表面结构,是从外膜突出的薄的丝状细胞表面突起,Porphyromonasgingivalis主要有长菌毛和短菌毛,其中牙周炎患者中分布最广的是I型和II长菌毛型。菌毛FimA和Mfa1均有利于Porphyromonasgingivalis早期黏附并定植到唾液蛋白、真核细胞、细胞外基质或其他细菌,并且能够增强生物膜形成、细菌运动、细菌与宿主细胞的黏附及细菌侵入细胞。长菌毛可以被上皮细胞、内皮细胞和免疫细胞的TLR2受体识别激活NF-κB通路并诱导促炎细胞因子和黏附分子的产生,而CD14是识别PorphyromonasgingivalisFimA所需的TLR2 共同受体,与TLR2受体共同作用[18]。长菌毛与单核细胞/巨噬细胞中的补体受体3(complement receptors,CR3;CD11b/CD18) 相互作用,导致细胞外信号调节激酶 1/2磷酸化并抑制 LPS 诱导的白细胞介素-12(interleukin cell-12,IL-12)产生。短菌毛除了与 TLR2 和 CD14 相互作用诱导细胞因子产生外,还可刺激树突状细胞吞噬Porphyromonasgingivalis[19],但促进了抗菌自噬和溶酶体融合的逃逸,导致Porphyromonasgingivalis在骨髓树突细胞中持续存在[20-22]。
1.5 荚膜
Porphyromonasgingivalis荚膜是Porphyromonasgingivalis的表面结构,是毒力因子之一,由氨基半乳糖、氨基葡萄糖等构成的黏附于细胞壁外层的多聚物。研究发现,在有荚膜和无荚膜的Porphyromonasgingivalis小鼠模型中有荚膜的Porphyromonasgingivalis毒力较强,能引起小鼠全身感染[3],并且发现感染有荚膜Porphyromonasgingivalis的小鼠产生IL-8 等炎症因子较少,可能与其抗吞噬,减缓伤口愈合有关[23]。Porphyromonasgingivalis的致病因子如图1所示。

图1 Porphyromonas gingivalis的致病因子
2 Porphyromonas gingivalis与心血管疾病关系
Porphyromonasgingivalis与心血管疾病密切相关,研究表明,患有牙周病的患者其患心血管疾病的风险更高[24]。CVD包括心肌梗死(myocardial infarction,MI)、高血压、心力衰竭、动脉粥样硬化(atherosclerosis,AS)、心肌炎、冠心病等[25]。报道中显示129 630名志愿者对22项研究报告表明牙周炎患者患心肌梗死的概率比非牙周炎患者高出2倍[26]。在许多动脉粥样硬化术后病灶标本中检出了高含量的Porphyromonasgingivalis[27-29]。64.1%的主动脉瘤患者的动脉瘤标本和口腔牙菌斑中发现Porphyromonasgingivalis的感染等[30-31]。Porphyromonasgingivalis与CVD相关性研究,证实了Porphyromonasgingivalis从多个途径感染CVD相应病灶部位,它们之间存在着相关性(表1)。

表1 Porphyromonas gingivalis与CVD相关性研究Table 1 Study on the correlation between Porphyromonas gingivalis and CVD
3 Porphyromonas gingivalis对CVD的致病途径
Porphyromonasgingivalis对CVD的致病途径主要为吞噬细胞介导和通过入血导致菌血症引起心血管疾病。
3.1 吞噬细胞介导
在CVD中,血管内皮受到异常刺激,导致血小板黏附在内皮细胞脱落的区域[39]。并从血液中招募单核细胞黏附到内膜下并持续释放前炎性介质,最终分化为泡沫细胞和导致VSMCs增生[40],从而形成成熟的斑块并增大,甚至阻塞血管导致CVD发生[41-43]。
3.2 菌血症感染
慢性牙周炎有高达8~20 cm2的牙周袋溃疡上皮衬里,如果损伤使牙周袋溃疡上皮衬里连续性中断,则有可能导致Porphyromonasgingivalis进入血液循环引起菌血症[44]。从而导致血管内皮的通透性被破坏,低密度脂蛋白(density lipoprotein,LDL)被VSMCs摄入,同时氧化应激发生在内皮中并产生大量超氧化物,形成泡沫细胞,然后被氧化得到氧化低密度脂蛋白(Oxidized density lipoprotein,oxLDL),导致VSMCs增生,从而导致CVD发生[45]。Porphyromonasgingivalis进入心血管系统的致病途径如图2所示。
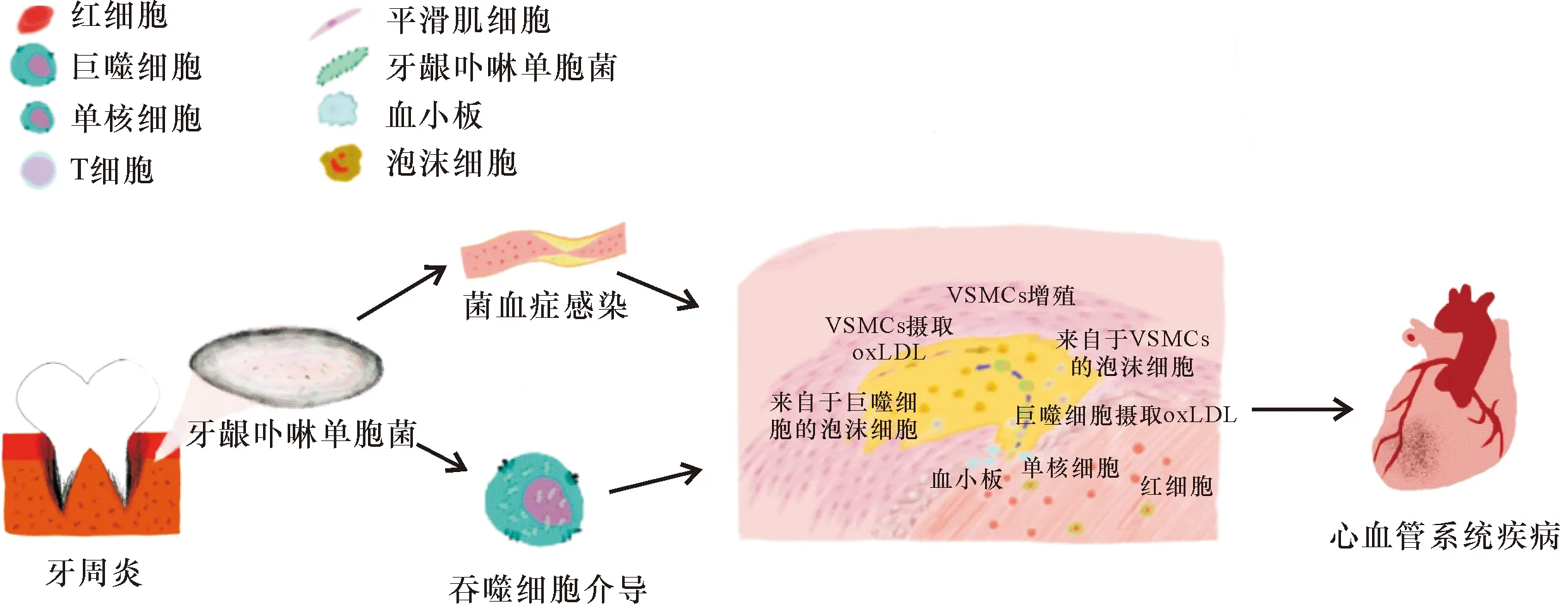
图2 Porphyromonas gingivalis进入心血管系统的致病途径
4 Porphyromonas gingivalis对CVD的致病机制
在机制上,Porphyromonasgingivalis及其释放的毒性产物可通过引发机体一系列的免疫炎症反应,使得炎症介质CRP、IL-6、TNF-α等进入血循环,从而影响心血管疾病的发生和发展[46]。Porphyromonasgingivalis对CVD的致病机制有以下几个方面:氧化应激(oxidative Stress,OS)和内皮功能障碍;脂质蓄积与泡沫细胞形成;血管重塑;血管钙化;血栓形成;斑块破裂。
4.1 氧化应激和内皮功能障碍
OS是体内氧化与抗氧化失衡的一种状态,其产生的中间产物对体内组织产生不良影响。Shiheido等[2]研究发现,Porphyromonasgingivalis侵入了 MI 小鼠的缺血心肌,增加氧化应激反应,基质金属蛋白酶-9(matrix met-alloproteinases,MMP-9)活性增加,并诱导心脏破裂。Xie等[29]发现,Porphyromonasgingivalis感染可激发TLRs-NF-κB信号转导轴和增加DNA甲基转移酶1(DNA methyltransferase 1,DNMT-1)表达,使钟基因BMAL1的转录下降,并增强钟基因CLOCK表达使p65磷酸化,使NF-κB信号传导增加,促进氧化应激和人主动脉内皮细胞的炎症反应,从而诱导AS发生。内皮功能障碍是内皮在血管疾病中的全身病理状态[47],Porphyromonasgingivalis可通过改变炎症物质和增加血管紧张素Ⅱ从而使血管内皮功能障碍诱导AS、高血压发生[1]。Sampath等[47]的研究发现,Porphyromonasgingivalis感染的原代人主动脉内皮细胞活力下降,诱导二氢叶酸还原酶mRNA下降,糖原合酶激酶3β的mRNA增高,从而使血管内皮功能障碍,诱导AS等其他心血管疾病发生。因此,在慢性牙周炎中,Porphyromonasgingivalis可诱导诱发氧化应激反应,并且介导巨噬细胞摄取胆固醇和转变成泡沫细胞[48],促进促炎细胞因子释放、通过NF-κB、NF-κB-BMAL1-NF-κB、NLRP3、激活蛋白 1 (AP-1)、p38、c-Jun N-激酶等通路增加血管内皮损伤[29,49-50]。Porphyromonasgingivalis诱导血管内皮细胞间质转化和凋亡,损害其完整性并失去修复能力[51],证实其在破坏心血管内皮稳态的进展中产生关键作用[52-54]。因此,在实验设计和临床研究中,如果能从减缓Porphyromonasgingivalis介导的炎症因子的释放和氧化应激反应而增加血管内皮细胞的完整性和增强其修复功能的切入点进行研究,或许能减少心血管疾病的发生发展。
3)验证性实验过多,不能培养学生的实践能力。所使用的实验设备为模拟电路工具箱,学生实验只是连接封装的模型,由示波器观察实验结果。实验内容包含典型环节模拟、二阶系统时间响应、典型环节频率特性以及控制系统分析,这些实验均以验证性为主,延展性不足,无法达到锻炼动手能力的目的,对于学生理解帮助不大。
4.2 脂质蓄积与泡沫细胞形成
LDL通过氧化修饰激活内皮等细胞,其细胞因子能够使血液循环中的单核细胞迁移到内膜并分化成巨噬细胞,并衍生出泡沫细胞,而Porphyromonasgingivalis能够增加VSMCs摄取脂质并在巨噬细胞内蓄积[55]。Yang等[56]研究表明Porphyromonasgingivalis通过LDL诱导泡沫细胞形成并溶酶体整合膜蛋白2(Lysosomal integrated membrane protein 2,LIMP2)的表达导致动脉壁中脂质沉积和斑块形成[57],内膜细胞增生,血流流动受阻,诱发心血管疾病[56,58]。同时发现Porphyromonasgingivalis促进泡沫细胞的形成,并使溶酶体整合膜蛋2(lysosomal integral membrane protein 2,LIMP2)表达上升、NF-κB通路激活和JNK激酶(Jun kinas,JNK)活性升高,使Porphyromonasgingivalis诱导的LIMP2 的表达升高,导致泡沫细胞形成,诱导AS发生。
这些研究表明,Porphyromonasgingivalis-LPS可干扰巨噬细胞中的脂质代谢,增加巨噬细胞对LDL胆固醇的摄取,并通过LDL刺激巨噬细胞和泡沫细胞导致促炎细胞因子上调,从而诱导AS的发生发展。因此,如何通过减缓Porphyromonasgingivalis-LPS的作用来调控巨噬细胞对脂质的摄取代谢,减少泡沫细胞形成,或许能减少Porphyromonasgingivalis对CVD的加剧。
4.3 血管重塑
血管重塑是通过使血管壁增厚或变薄,从而改变血管动力学,是一种病理性的适应性改变,Porphyromonasgingivalis可增加血管斑块的形成,促使血管重塑,防止血管变窄发生缺血。DeLeon-Pennell等[32]发现,接种Porphyromonasgingivalis-LPS的小鼠分泌Ccl12促炎因子增多,修复反应减少,疤痕形成减少,单核细胞趋化蛋1(monocyte chemoattractant protein 1,MCP-1)增多,致使血管重塑,从而对MI后的左心室产生不良影响,诱导MI发生。Zaidi等[33]发现,Porphyromonasgingivalis-LPS激活记忆CD8+T细胞,巨噬细胞增多,导致MI后心脏血管重塑不良,从而诱导MI发生。
因此,为了适应血管内膜的增生、斑块增大,血管得以重塑,以维持恒定的血流防止缺血,Porphyromonasgingivalis释放的牙龈蛋白酶可刺激主动脉平滑肌细胞中血管生成素-2的表达,促使主动脉平滑肌细胞迁移诱导血管重塑。而Porphyromonasgingivalis-LPS可上调血管平滑肌VSMCs中内皮素β受体的表达,进而使动脉对内皮素-1介导的血管收缩敏感[59-60]。所以,如何从减缓Porphyromonasgingivalis及其毒性物质能抑制ECs的功能、增殖和迁移并诱导凋亡的角度出发,从而减少诱导血管重塑并加重CVD还有许多值得研究的地方。
4.4 血管钙化
血管钙化是血管壁出现异常的钙化沉积,从而使血管壁硬化,舒张收缩功能下降,引起局部缺血。唐路等[35]发现,Porphyromonasgingivalis诱导VSMCs活力和钙化增高,VSMCs异常的分化、迁移和凋亡会造成血管的损伤,导致主动脉壁钙化增高,导致血管钙化,诱导AS的发生。
血管壁的中内膜层,是血管发生钙化的结构基础,许多研究都表明血管钙化是由VSMCs分化所致[61]。而Porphyromonasgingivalis可诱导VSMCs活性增强并分化,并且导致VSMCs和主动脉钙化,并直接引起主动脉壁钙化沉积。血管钙化使血管硬度增加,斑块形成,并导致AS的发生[62-64]。临床上可加强牙周等治疗,改善口腔卫生环境,从而减少Porphyromonasgingivalis的产生而导致的血管钙化引起的AS。
4.5 血栓形成
Porphyromonasgingivalis释放的牙龈蛋白酶诱导ECs的促凝,并降低ECs的质膜上的血栓调节蛋白(CD141)的活性,使转化为抗凝酶的凝血酶可以与血小板结合,刺激血小板聚集,形成血栓[53,65]。Senini等[66]研究发现,Porphyromonasgingivalis-LPS 刺激Cdc42升高,凝血时间减少,导致血栓形成,诱导MI等心血管疾病的发生。为防止失血和维持血管的完整性,血管损伤后血栓便形成了,这不仅减少了血流量,而且导致了组织缺氧和梗死,诱导并加重MI。
4.6 斑块破裂
Zhang等[53]研究发现,Porphyromonasgingivalis增多可导致辅助性T 细胞17(T helper cell 17,Th17)增多和调节性T细胞(Regulatory cells,Tregs)减少,导致Th17/Tregs失衡和斑块不稳定。Porphyromonasgingivalis及代谢产物使单核细胞增多,诱导Th17/IL-17 反应增多,使 TNF-α、IL-1β、IL-6 和 IL-17升高,通过 TLR2/TLR4 调控和炎症反应形成AS斑块,导致AS发生。
5 小结与展望
综上所述,口腔微生物的发生发展与心血管系统疾病的发生息息相关,Porphyromonasgingivalis是其中常见而典型的细菌,研究其中的关系将为未来口腔与心血管疾病的防治提供重要参考价值,有利于疾病的转归。
在牙周炎致病过程中,Porphyromonasgingivalis是其常见致病菌,当合并其他细菌如伴放线杆菌(Aa)、福赛坦氏菌等时,它们的致病因子、致病途径和致病机制对CVD的影响值得深入探讨。
在致病因子上,Porphyromonasgingivalis的菌毛、血凝素均有不同分型,还有牙龈素、Toll样受体、胶原酶等不常见的毒力因子未被深入研究,它们可对Porphyromonasgingivalis的毒力、侵袭力产生影响并可通过调节宿主的免疫、炎症等反应,诱导心血管系统疾病的发生发展,可以对其做探讨。
在发病机制上,目前许多报道Porphyromonasgingivalis与CVD的密切关系,Porphyromonasgingivalis可通过募集黏附单核细胞,使泡沫细胞形成和内膜细胞增生,促进巨噬细胞表面LDL增加导致硬化斑块及血栓形成,从而引发CVD[68]。然而,对于急性心肌梗死(AMI),Sun等[61]发现Porphyromonasgingivalis毒素虽可激活记忆 CD8+T 细胞,但在阻断CD8+T 细胞的激活后,并没有影响巨噬细胞数量或使其细胞壁变薄,表明该激活CD8+T 细胞可能只是AMI的发生其中一个原因,其原理值得深究[33];另一方面,Porphyromonasgingivalis对血管钙化的作用研究目前也较少;因此,Porphyromonasgingivalis参与CVD发生发展的过程及发病机制尚有许多值得探讨与深入研究的地方。
在治疗上,Shiheido等[2]用免疫球蛋白 Y 对抗牙龈蛋白酶治疗显著降低了由心脏破裂引起的 Pg 接种 MI 小鼠的死亡率;Chen 等[69]的发现表明BMP4 在感染相关血管钙化中可能具有治疗作用。唐路等[35]发现通过牙周治疗,可减缓颈总动脉内膜厚度增加的进程。这都是我们在临床上治疗思路值得借鉴的地方,通过有效牙周治疗,减少Porphyromonasgingivalis等炎症因子,有利于防治CVD。也可以通过增加临床样本量,对大量受试者进行随访,明确牙周治疗对CVD转归的影响。
作者贡献声明
胡颖文:提出研究思路,整理文献,撰写论文;马晓岚:提出研究框架,修改论文;牛海涛:制定大纲,审核论文;黄跃:提出研究思路,修改论文。
利益冲突声明
本研究未受到企业、公司等第三方资助,不存在潜在利益冲突。
猜你喜欢
中国生殖健康(2019年6期)2019-01-06
文苑(2018年22期)2018-11-19
知识就是力量(2018年6期)2018-06-15
小学生导刊(2018年4期)2018-04-18
益寿宝典(2017年36期)2017-08-20
新农业(2016年18期)2016-08-16
四川畜牧兽医(2016年1期)2016-04-05
西南军医(2016年6期)2016-01-23
食品工业科技(2014年7期)2014-03-11
中国预防兽医学报(2013年10期)2013-09-10
