
双杂环基甲酮类化合物的设计、合成及杀线虫活性
2023-06-20 06:19曹晓峰徐晓勇
农药学学报 2023年3期
邱 蝶, 曹晓峰, 李 忠, 徐晓勇
(华东理工大学 药学院 上海市化学生物学 (芳香杂环) 重点实验室,上海 200237)
植物寄生线虫对农作物的健康生长构成了巨大威胁,严重影响作物产量,其造成的经济损失占全球作物产量损失的14%,每年约1730 亿美元[1-2],其中根结线虫(Meloidogynespp.)种类繁多,能侵染多种作物的根系,造成的农业损失也最为显著[3-4]。目前世界各地已使用多种化学杀线虫剂控制线虫,然而大多数化学杀线虫剂作用谱广、选择性差、对有益土壤微生物和环境产生了不利影响,正越来越多地被停用或被淘汰[5-6],只有少数几个新开发的杀线虫剂安全性较好[7-8],如氟噻虫砜[9]、tioxazafen[10]、氟吡菌酰胺[11]、三氟咪啶酰胺[12]、cyclobutrifluram[13]和三氟杀线酯[14]等。因此,亟待开发更多新型高效、低毒、低残留和环境兼容性好的杀线虫剂。
稠环的优化策略在先导发现的药物分子设计中十分常见,一般是将稠环部分变为单环。例如,拜耳对杜邦推出的三氟咪啶酰胺的优化就采用了类似的策略,在保留苯基磺酰基酰胺结构的基础上,拜耳的研究人员通过将吡啶并咪唑修改为咪唑或吡唑,并引入不同类型结构片段,得到了化合物P1与P2(图式1),这两个化合物均展现出对线虫良好的杀虫活性,其中化合物P2在0.03125 mg/L 下对马铃薯胞囊线虫Heterodera pallida的抑制率达到90%[15-16]。此外,苯环 (脂肪环) 连杂环化合物和杂环连杂环化合物也被广泛应用于药物分子设计中,如今已有许多此类农药登记上市,例如杀线虫剂tioxazafen、杀虫剂nicofluprole[17]、isocycloseram[18-19]和tyclopyrazoflor[20]、杀菌剂pyridachlometyl[21]和唑菌酯 (pyraoxystrobin)[22]以及除草剂氯氟吡啶酯 (florpyrauxifen-benzyl)[23]、methiozolin[24]和氯丙嘧啶酸 (aminocyclopyrachlor)[25]等 (图式2)。

图式1 三氟咪啶酰胺的稠环优化策略Scheme 1 Fused ring optimization strategy of fluazaindolizine
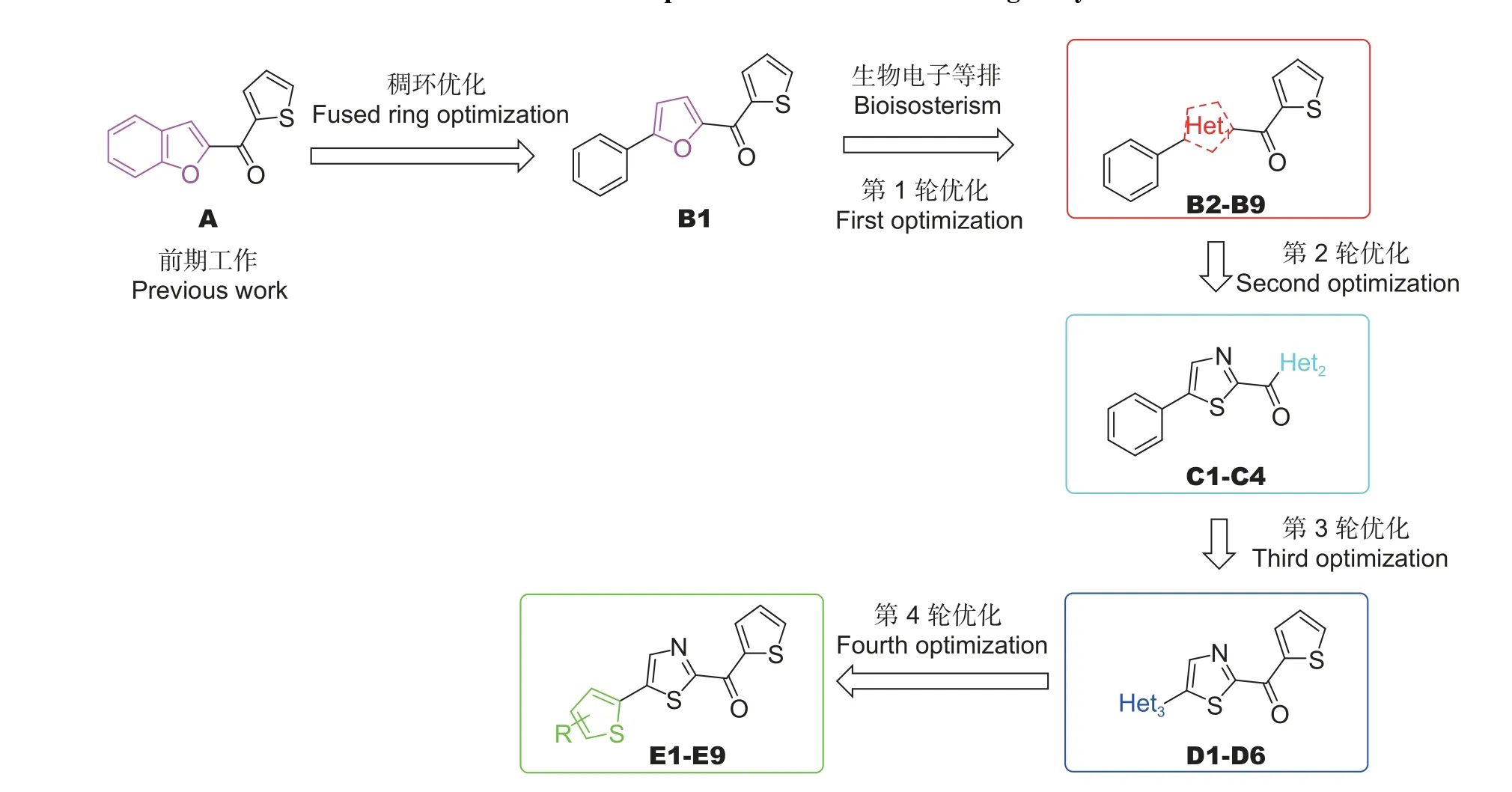
本课题组在先前的工作中得到了高活性化合物A(图式3) ,其在离体条件下在质量浓度为40 mg/L 时对南方根结线虫的致死率为85.68%,在5 mg/L 下,其在沙土中因南方根结线虫造成的根结抑制率为81.39%,在40 mg/L 下,其在基质中的根结抑制率为53.09%[26]。为了继续探究对南方根结线虫具有更高活性的化合物,本文采用类似于三氟咪啶酰胺的稠环优化策略,将化合物A中苯并呋喃的稠环部分变换成苯连呋喃,得到了先导化合物B1,并通过生物电子等排设计合成了27 个未见文献报道的化合物。目标化合物的优化策略见图式3,合成路线见图式4。
1 材料与方法
1.1 仪器与试剂

图式2 双环相连结构的商品化农药品种Scheme 2 Commercialized pesticides varieties containing biscyclo structure
图式3 目标化合物的分子设计Scheme 3 Molecular design of target compounds
Bruker AM-400 核磁共振分析仪 (德国Bruker公司,以CDCl3或者DMSO-d6为溶剂,TMS 为内标);Buchi Melting Point B-540 熔点仪(瑞士Buchi 公司),未经校正;MIicroMass GCT CA055高分辨质谱仪 (HRMS-EI-TOF,美国Waters 公司);ThermoFisher Q-Exactive (ESI-Orbitrap) 液相色谱-质谱联用仪 (中国赛默飞世尔科技公司);Agilent 7890A/5975C 气质联用仪 (GC-MS,美国惠普公司)。
本文试验所用的试剂及溶剂均为分析纯或市售分析纯。对照药剂为99%噻唑膦 (fosthiazate)原药,从湖北常鑫盛化工有限公司购买。
1.2 化合物的合成
1.2.1 中间体的合成

图式4 目标化合物的合成路线Scheme 4 Synthetic routes of target compounds
1.2.1.1 中间体1~3 的合成 参考文献方法[27]合成中间体1。向100 mL 圆底烧瓶中依次加入40 mL 1,4-二氧六环、4 mL 水、碳酸钾 (10 mmol)、苯硼酸 (6 mmol)、5-溴-2-糠酸甲酯/5-溴噻吩-2-甲酸甲酯 (5 mmol) 和四三苯基磷钯 (0.05 mmol),氩气置换,搅拌加热至100 ℃,反应6 h 后,薄层色谱(TLC) 跟踪反应进程 (V(石油醚) :V(乙酸乙酯) =3 : 1)。待反应完毕将反应液倒入硅藻土中抽滤,并用二氯甲烷 (20 mL × 2) 洗涤滤饼,减压浓缩。用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,得到粗产物1。
参考文献方法[27]合成中间体2。向100 mL 圆底烧瓶中依次加入10 mL 浓度为2 mol/L 的氢氧化钠溶液、30 mL 甲醇和中间体1,搅拌加热至65 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 1 : 1) 跟踪反应进程。待反应完毕,减压浓缩,向烧瓶中加入20 mL 水,室温下搅拌,缓慢加入1 mol/L 的盐酸溶液使溶液pH = 5,搅拌4 h 后抽滤得到中间体2,产率55%。X 为O 时:1H NMR (400 MHz,DMSO-d6),δ:7.80~7.69 (m, 2H), 7.50~7.41 (m,2H), 7.37~7.29 (m, 1H), 7.02~6.95 (m, 2H).X 为S 时:1H NMR (400 MHz, DMSO-d6),δ: 7.71 (d,J=7.4 Hz, 2H), 7.57 (d,J= 4.0 Hz, 1H), 7.49 (d,J= 4.0 Hz,1H), 7.47~7.42 (m, 2H), 7.41~7.31 (m, 1H)。
参考文献方法[28]合成中间体3。向100 mL 圆底烧瓶中依次加入35 mL 甲苯、2-氯-4,6-二甲氧基-1,3,5-三嗪 (CDMT) (10 mmol) 和N-甲基吗啡啉(10 mmol),室温下搅拌,将15 mL 甲苯与对应的中间体2 (10 mmol) 依次加入恒压滴液漏斗并滴加,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪反应进程。反应结束后,减压浓缩,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,得到粗产物中间体3。
1.2.1.2 中间体4~7、10~14 的合成 参考文献方法[29-30]合成中间体4~6。向100 mL 圆底烧瓶中依次加入35 mL 二氯甲烷、对应的2-氨基苯乙酮盐酸盐 (2 mmol) 和三乙胺 (4.8 mmol),室温下搅拌,将10 mL 二氯甲烷和2-噻吩乙酰氯 (2.4 mmol)依次加入恒压滴液漏斗中并滴加,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪至反应结束。向烧瓶中加入适量的饱和氯化钠水溶液,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,用无水硫酸钠干燥,减压浓缩,得到粗品中间体4。向50 mL圆底烧瓶中依次加入15 mL 三氯氧磷和粗产物中间体4 (2 mmol),氩气置换,搅拌加热至107 ℃,反应4 h 后,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1)跟踪至反应完毕。将反应液缓慢加入饱和碳酸氢钠的冰水溶液中,用乙酸乙酯萃取 (20 mL × 3),收集上层有机相,使用无水硫酸钠干燥,减压浓缩,硅胶柱层析分离提纯得到中间体5,产率62%。1H NMR (400 MHz, DMSO-d6),δ: 7.69~7.65(m, 2H), 7.60 (s, 1H), 7.50~7.41 (m, 3H), 7.38~7.33(m, 1H), 7.06 (dd,J= 3.2, 1.0 Hz, 1H), 7.00 (dd,J=5.0, 3.2 Hz, 1H), 4.45 (s, 2H)。向100 mL 圆底烧瓶中依次加入40 mL 甲苯、中间体4 (2 mmol) 和劳森试剂 (3.0 mmol),搅拌加热至110 ℃,TLC(V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪反应进程,待反应完毕,减压浓缩,向烧瓶中加入适量的饱和氯化钠水溶液,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,硅胶柱层析分离提纯,得到中间体6,产率67%。1H NMR (400 MHz, CDCl3),δ: 7.88 (s, 1H),7.53 (d,J= 7.4 Hz, 2H), 7.43~7.36 (m, 1H),7.36~7.31 (m, 1H), 7.29~7.23 (m, 1H), 7.08~6.97 (m,2H), 4.57 (s, 2H)。
参考文献方法[31]合成中间体7。向50 mL 圆底烧瓶中依次加入15 mL 三氯氧磷、苯甲酰肼(2 mmol) 和2-噻吩乙酸 (2.4 mmol),氩气置换,搅拌加热至107 ℃,反应4 h 后,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪反应进程,待反应完毕,将反应液缓慢加入含饱和碳酸氢钠的冰水溶液中,用乙酸乙酯萃取 (20 mL × 3),收集上层有机相,使用无水硫酸钠干燥,减压浓缩,硅胶柱层析分离提纯得到中间体7,产率32%。1H NMR(400 MHz, CDCl3),δ: 7.98~7.91 (m, 2H), 7.48~7.35(m, 3H), 7.16 (dd,J= 5.2, 1.0 Hz, 1H), 6.96 (dd,J=3.2, 1.0 Hz, 1H), 6.90 (dd,J= 5.2, 3.2 Hz, 1H), 4.41(s, 2H)。
参考文献方法[32-33]合成中间体10~11。向100 mL圆底烧瓶中依次加入40 mL 乙醇、盐酸羟胺 (2.5 mmol)、N,N-二异丙基乙胺 (2.5 mmol) 和苯甲腈(2 mmol),搅拌加热至60 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪反应进程,待反应完毕,减压浓缩,得到粗品中间体10。向100 mL圆底烧瓶中依次加入40 mL1,4-二氧六环、N-羟基苯甲酰胺 (2 mmol) 和N,N-二异丙基乙胺 (3 mmol),室温下搅拌,将10 mL1,4-二氧六环和2-噻吩乙酰氯 (2 mmol) 依次加入恒压滴液漏斗中并滴加,原料消失后室温下额外搅拌1 h,随后反应加热至100 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪反应进程,待反应完毕,减压浓缩,向烧瓶中加入适量的饱和氯化钠水溶液,用二氯甲烷萃取(20 mL × 3),收集上层有机相,使用无水硫酸钠干燥,减压浓缩,硅胶柱层析分离提纯得到中间体11,产率45%。1H NMR (400 MHz, DMSO-d6),δ: 8.03~7.97 (m, 2H), 7.64~7.53 (m, 3H), 7.50 (dd,J= 5.2, 1.2 Hz, 1H), 7.14 (dd,J= 3.6, 1.2 Hz, 1H),7.03 (dd,J= 5.2, 3.6 Hz, 1H), 4.72 (s, 2H)。
中间体12 的合成步骤与中间体10 相同,产率45%;中间体13 的合成步骤与中间体11 相同,产率40%。中间体13:1H NMR (400 MHz,CDCl3),δ: 8.03~7.94 (m, 2H), 7.47~7.30 (m, 3H),7.16 (dd,J= 5.2, 1.0 Hz, 1H), 6.97 (dd,J= 3.4, 1.0 Hz, 1H), 6.89 (dd,J= 5.2, 3.4 Hz, 1H), 4.41 (s,2H)。
参考文献方法[34]合成中间体14。向100 mL圆底烧瓶中依次加入40 mL 的N,N-二甲基甲酰胺(DMF)、2-(2-噻吩基)硫代乙酰胺 (1 mmol) 和溴代苯乙酮 (1.2 mmol),搅拌加热至90 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪反应进程,向烧瓶中加入适量的饱和氯化钠水溶液,待反应完毕,用乙酸乙酯萃取 (20 mL × 3),收集上层有机相,使用无水硫酸钠干燥,减压浓缩,硅胶柱层析分离提纯,得到中间体14,产率75%。1H NMR(400 MHz, CDCl3),δ: 7.87~7.78 (m, 2H), 7.80~7.71(m, 3H), 7.36 (s, 1H), 7.19~7.15 (m, 1H), 6.97~6.95(m, 1H), 6.94~6.89 (m, 1H), 4.52 (s, 2H)。
1.2.1.3 中间体16-1、16-2、16-3、16 的合成 向250 mL 圆底烧瓶中依次加入40 mL 二氯甲烷、2-氨基苯乙酮盐酸盐 (40 mmol) 和三乙胺 (96 mmol),冰浴下搅拌,将10 mL 二氯甲烷和草酰氯单乙酯(48 mmol) 依次加入恒压滴液漏斗中并滴加,滴加完成后转至室温下搅拌,TLC (V(石油醚) :V(乙酸乙酯) = 3 : 1) 跟踪至反应结束。向烧瓶中加入适量饱和氯化钠水溶液,使用二氯甲烷萃取 (20 mL ×3),收集下层有机相,使用无水硫酸钠干燥,减压除去二氯甲烷,硅胶柱层析分离提纯,得到中间体16-1,产率30%。1H NMR (400 MHz, CDCl3),δ: 8.10 (s, 1H), 8.00 (d,J= 7.4 Hz, 2H), 7.65 (t,J=7.4 Hz, 1H), 7.52 (t,J= 7.8 Hz, 2H), 4.84 (d,J=4.8 Hz, 2H), 4.40 (q,J= 7.2 Hz, 2H), 1.41 (t,J= 7.2 Hz, 3H)。
向100 mL 圆底烧瓶中依次加入30 mL1,2-二氯乙烷、中间体16-1 (9 mmol) 和劳森试剂 (10.8 mmol),搅拌加热至60 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪至反应结束后,停止加热。减压除去1,2-二氯乙烷,使用二氯甲烷萃取(20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压除去二氯甲烷,硅胶柱层析分离提纯,得到中间体16-2,产率88%。1H NMR (400 MHz, CDCl3),δ: 8.16 (s, 1H), 7.63 (dd,J= 8.0, 1.4 Hz, 2H), 7.50~7.38 (m, 3H), 4.50 (q,J= 7.2 Hz, 2H),1.46 (t,J= 7.2 Hz, 3H)。
向100 mL 圆底烧瓶中依次加入10 mL 浓度为2 mol/L 的氢氧化锂水溶液、30 mL1,4-二氧六环和中间体16-2,室温搅拌,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪反应进程。待反应完毕,减压浓缩除去1,4-二氧六环。向烧瓶中加入20 mL水,室温下搅拌,缓慢加入2 mol/L 的盐酸溶液使溶液pH = 2。搅拌4 h 后,使用二氯甲烷萃取(20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压除去二氯甲烷,得到中间体16-3,产率50%。1H NMR (400 MHz, DMSO-d6),δ: 9.10 (s,1H), 8.33 (s, 1H), 7.74~7.67 (m, 2H), 7.50~7.43 (m,2H), 7.42~7.35 (m, 1H)。
向100 mL 圆底烧瓶中依次加入30 mL 二氯甲烷、中间体16-3 (3 mmol) 和0.1 mL DMF,冰浴下搅拌,将10 mL 二氯甲烷和草酰氯 (12 mmol)依次加入恒压滴液漏斗中并滴加,TLC (V(石油醚) :V(乙酸乙酯) = 10 : 1) 跟踪至反应完毕。减压浓缩除去二氯甲烷,得到中间体16。
1.2.1.4 中间体17-1、17-2 的合成 向100 mL 圆底烧瓶中依次加入50 mL 乙酸乙酯、2-乙酰基芳杂环 (10 mmol) 与溴化铜 (12 mmol),搅拌加热至77 ℃,通过气相色谱-质谱 (GC-MS) 跟踪反应。反应结束后,用乙酸乙酯萃取 (20 mL × 3),用饱和氯化钠水溶液洗涤,合并有机相使用无水硫酸钠干燥,减压浓缩得到中间体17-1。
向100 mL 圆底烧瓶中依次加入40 mL 乙腈、中间体17-1 (2 mmol) 和二甲酰胺钠 (2.4 mmol),加热至81 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1)跟踪至反应结束。减压浓缩,用二氯甲烷萃取(20 mL × 3),加入饱和氯化钠水溶液洗涤,合并有机相使用无水硫酸钠干燥,减压浓缩。将所得残留物与30 mL 浓度为5 mol/L 的盐酸一起加入100 mL 圆底烧瓶中,加热至100 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 3 : 1) 跟踪反应进程,反应结束后使用二氯甲烷萃取 (20 mL × 2),加入去离子水溶液洗涤,合并水相,减压浓缩得到中间体17-2。
1.2.1.5 中间体18 的合成 向100 mL 圆底烧瓶中依次加入30 mL 丙酮、中间体17 (3 mmol) 和高锰酸钾 (6 mmol),室温下搅拌,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪至反应完毕。将反应液使用硅藻土抽滤,并用二氯甲烷 (10 mL × 2) 洗涤滤饼,减压浓缩,柱层析分离,得到中间体18,产率48%。1H NMR (400 MHz, CDCl3),δ: 8.57 (d,J=3.8 Hz, 1H), 8.13 (s, 1H), 7.74 (d,J= 4.8 Hz, 1H),7.53 (d,J= 8.4 Hz, 2H), 7.46 (d,J= 8.4 Hz, 2H),7.17 (t,J= 4.4 Hz, 1H)。
1.2.1.6 中间体8~9、15-1、15、17-3、17 的合成
中间体8、15-1、17-3 的合成步骤与中间体4 相同,中间体9、15、17 的合成步骤与中间体6 相同。中间体9:1H NMR (400 MHz, CDCl3),δ:7.95~7.86 (m, 2H), 7.52~7.39 (m, 3H), 7.25 (dd,J=5.2, 1.2 Hz, 1H), 7.03 (dd,J= 3.4, 1.2 Hz, 1H), 6.99(dd,J= 5.2, 3.4 Hz, 1H), 4.68 (s, 2H)。
1.2.2 目标化合物的合成
1.2.2.1 目标化合物B1~B2 的合成 参考文献方法[28]合成。向100 mL 圆底烧瓶中依次加入30 mL甲苯、中间体3 (10 mmol)、2-噻吩硼酸 (12 mmol)、双三苯基磷二氯化钯 (0.2 mmol) 和磷酸钾 (40 mmol),氩气置换,搅拌加热至110 ℃,反应30 min,停止加热待反应冷却至室温,将反应液使用硅藻土抽滤,并用二氯甲烷 (10 mL × 2) 洗涤滤饼,合并有机相,减压浓缩,柱层析 (V(石油醚) :V(乙酸乙酯) = 5 : 1) 分离,得到化合物B1-B2。
1.2.2.2 目标化合物C1 和D2 的合成 向50 mL圆底烧瓶中依次加入10 mL 二甲基亚砜、芳杂环丙氨酸 (5 mmol)、九水合硫化钠 (3 mmol)、碘单质 (5 mmol) 和醋酸 (1.25 mmol),搅拌加热至100 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪反应进程,待反应完毕,向烧瓶中加入饱和硫代硫酸钠的水溶液淬灭,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,柱层析分离,得到化合物C1 和D2。
1.2.2.3 目标化合物C3 的合成 向100 mL 圆底烧瓶中依次加入30 mL 无水二氯甲烷和中间体20(5 mmol),冰浴下搅拌30 min,将10 mL 无水二氯甲烷和吡咯 (5 mmol) 依次加入恒压滴液漏斗中并滴加,滴加完成后转至室温下搅拌,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪至反应完毕。加入冰水和1 mol/L 盐酸淬灭,淬灭完成后将反应液使用硅藻土抽滤,并用二氯甲烷 (10 mL × 2) 洗涤滤饼,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,柱层析分离,得到化合物C3。
1.2.2.4 目标化合物D6 的合成 向100 mL 圆底烧瓶中依次加入30 mL DMF、中间体18 (5 mmol)、4-甲苯硫酚 (10 mmol)、碳酸铯 (10 mmol) 和碘化亚铜 (1 mmol),搅拌加热至90 ℃,TLC (V(石油醚) :V(乙酸乙酯) = 5 : 1) 跟踪至反应反应。将反应液使用硅藻土抽滤,并用二氯甲烷 (10 mL × 2)洗涤滤饼,用二氯甲烷萃取 (20 mL × 3),收集下层有机相,使用无水硫酸钠干燥,减压浓缩,柱层析分离,得到化合物D6。
1.2.2.5 目标化合物B3~B9、C2、C4、D1、D3~D5、E1~E9 的合成 合成步骤与中间体18 相同。
1.3 杀线虫活性测试
所有试验所用的南方根结线虫2 龄幼虫(J2),均由中国科学院上海生命科学研究院湖州现代农业生物技术产业创新中心培养。1.3.1 离体活性测试方法 参考文献方法[35]进行。将供试化合物溶解于有机溶剂 (丙酮或二甲基亚砜) 中,配制成质量浓度为1 × 104mg/L 母液,继而使用含曲拉通 (体积占比不超过0.15%) 的水溶液稀释,配制为待测药液。以噻唑膦 (fosthiazate)作为阳性对照,不含药物仅含有机溶剂的曲拉通水溶液作为空白对照。向96 孔板中加入所配制的药液,每孔加入50 μL (每个化合物每个浓度平行3 组),随后加入50 μL 含南方根结线虫2 龄幼虫悬浮液 (线虫约100~150 条),加盖,置于温度为25 ℃的避光环境中,分别于24、48 和72 h 使用体视显微镜观察统计死亡线虫数,于72 h 统计线虫死亡数与总数,并分别按 (1) 式和 (2) 式计算化合物对线虫的死亡率和校正死亡率。
(1) 式中:M0为死亡率,N1为孔板中死亡线虫数,N2为孔板中线虫总数;(2) 式中:M为校正死亡率,M1为处理组死亡率,M2为空白对照组死亡率。1.3.2 活体活性测试方法 参考文献方法[35],分别测试化合物在沙土和基质土壤中对因南方根结线虫造成的植物根结的抑制率。其中供试沙土为洗净的河沙;基质土壤为浙江湖州地区的红壤(组成成分为沙子、腐殖土和细土壤,对应的体积比为2 : 1 : 1)。
测试药液配制方法与1.3.1 节相同,有机溶剂为丙酮。使用噻唑膦作为阳性对照,仅含有机溶剂的曲拉通水溶液作为阴性组,仅加入线虫虫卵及纯水为空白对照。化合物在沙土中的测试方法与基质土壤中的测试方法相同。将1 周龄黄瓜幼苗种植于装有沙土或基质土壤的试管中,向每个试管中依次加入3 mL 的药液与3 mL 约含有2000 个南方根结线虫虫卵的悬浮液 (每个化合物每个浓度平行4 组),将试管置于20~25 ℃环境中,每天光照10 h、浇水,21 d 后将黄瓜幼苗从沙土或基质土壤中拔出,统计每株黄瓜根系上的根结数,按 (3) 式计算药物对根结的平均抑制率。
(3) 式中:根结抑制率I,阴性对照根结数S1,处理组根结数S2。
2 结果与讨论
2.1 目标的化合物合成
在合成目标化合物的过程中,一共进行了4 轮优化。第1 轮优化时,首先将先导化合物B1的芳香环部分用杂环进行替换,设计合成了化合物B2~B9;第2 轮优化时,基于B2~B9 中选出的化合物,将呋喃环更换为噻唑环,并对噻吩环部分做了生物电子等排,设计合成了化合物C1~C4;第3 轮优化时,保留2-噻唑(2-噻吩) 甲酮部分,用杂环替换结构中的苯环,设计合成了化合物D1~D6;第4 轮优化时,固定活性结构为噻吩-2-基(5-(噻吩-2-基))噻唑-2-基)甲酮,对噻吩环上的取代基进行变换,最终设计合成化合物E1~E9。化合物结构鉴定相关谱图数据如下。
(5-苯基-2-呋喃基)-2-噻吩甲酮 (B1): 白色固体,产率67%,m.p.94.5~95.3 ℃;1H NMR (400 MHz, DMSO-d6),δ:8.28 (dd,J= 3.8, 1.2 Hz, 1H), 8.14 (dd,J= 4.8, 1.2 Hz, 1H),7.98~7.91 (m, 2H), 7.68 (d,J= 3.8 Hz, 1H), 7.58~7.51 (m,2H), 7.50~7.43 (m, 1H), 7.37 (dd,J= 4.8, 3.8 Hz, 1H), 7.34 (d,J= 3.8 Hz, 1H).13C NMR (100 MHz, DMSO-d6),δ: 172.0,157.3, 150.5, 141.5, 135.2, 133.9, 129.4, 129.2, 129.0, 128.8,124.8, 121.7, 108.9.HRMS (EI): C15H10O2S (M+),计算值254.0402,测试值254.0400。
(5-苯基-2-噻吩基)-2-噻吩甲酮 (B2): 白色固体,产率63%,m.p.126.4~127.9 ℃;1H NMR (400 MHz, DMSO-d6),δ: 8.13~8.10 (m, 2H), 8.08 (d,J= 4.0 Hz, 1H), 7.83 (d,J= 7.2 Hz, 2H), 7.72 (d,J= 4.0 Hz, 1H), 7.53~7.47 (m, 2H),7.47~7.41 (m, 1H), 7.38~7.31 (m, 1H).13C NMR (100 MHz,DMSO-d6),δ: 177.6, 151.5, 141.9, 140.8, 135.1, 135.0, 133.7,132.5, 129.3, 129.2, 128.9, 126.1, 125.2 ppm.HRMS (EI):C15H10OS2(M+),计算值270.0173,测试值270.0172。
(5-苯基-2-噻唑基)-2-噻吩甲酮 (B5): 黄褐色固体,产率45%,m.p.142.5~143.3 ℃;1H NMR (400 MHz, CDCl3),δ: 8.64 (dd,J= 3.8, 1.2 Hz, 1H), 8.22 (s, 1H), 7.80 (dd,J=5.0, 1.2 Hz, 1H), 7.70~7.64 (m, 2H), 7.49~7.39 (m, 3H), 7.24(dd,J= 5.0, 3.8 Hz, 1H).13C NMR (100 MHz, CDCl3),δ:175.8, 165.6, 146.8, 140.3, 139.8, 136.8, 136.3, 130.6, 129.7,129.4, 128.4, 127.3.HRMS (EI): C14H9NOS2(M+),计算值271.0126,测试值 271.0129。
(5-苯基-1,3,4-噻二唑-2-基)-2-噻吩甲酮 (B6): 白色固体,产率51%,m.p.173.2~174.1 ℃;1H NMR (400 MHz,DMSO-d6),δ: 8.64 (dd,J= 4.0, 1.2 Hz, 1H), 8.31 (dd,J= 4.8,1.2 Hz, 1H), 8.21~8.09 (m, 2H), 7.71~7.57 (m, 3H), 7.42 (dd,J= 4.8, 4.0 Hz, 1H).13C NMR (100 MHz, DMSO-d6),δ:174.6, 172.1, 168.2, 138.9, 138.8, 137.7, 132.4, 129.6, 129.3,128.8, 128.3.HRMS (EI): C13H8N2OS2(M+),计算值272.0078,测试值272.0081。
(4-苯基-2-噻唑基)-2-噻吩甲酮 (B9): 粉色固体,产率58%,m.p.133.5~134.0 ℃;1H NMR (400 MHz, CDCl3),δ:8.62 (dd,J= 4.0, 1.2 Hz, 1H), 7.98~7.91 (m, 2H), 7.79 (s, 1H),7.76 (dd,J= 4.8, 1.2 Hz, 1H), 7.47~7.39 (m, 2H), 7.37~7.30(m, 1H), 7.19 (dd,J= 4.8, 4.0 Hz, 1H).13C NMR (100 MHz,CDCl3),δ: 175.7, 166.7, 157.5, 139.5, 136.9, 136.7, 133.7,129.0, 128.9, 128.3, 126.5, 119.8.HRMS (EI): C14H9NOS2(M+),计算值271.0126,测试值271.0129。
(5-苯基-2-噻唑基)-2-苯基甲酮 (C1): 褐色固体,产率:45%m.p.122.9~123.3 ℃;1H NMR (400 MHz, CDCl3),δ:8.44~8.36 (m, 2H), 8.16 (s, 1H), 7.65~7.54 (m, 3H), 7.51~7.44(m, 2H), 7.43~7.33 (m, 3H).13C NMR (100 MHz, CDCl3),δ:184.17, 166.29, 146.81, 140.38, 135.24, 133.51, 131.00,130.55, 129.60, 129.37, 128.41, 127.30.HRMS (EI):C16H11NOS (M+),计算值265.0561,测试值265.0563。
(5-苯基-2-噻唑基)-2-呋喃甲酮 (C2): 褐色固体,产率42%,m.p.119.8~120.8 ℃;1H NMR (400 MHz, DMSO-d6),δ: 8.61 (s, 1H), 8.23 (s, 1H), 8.18 (d,J= 3.6 Hz, 1H), 7.84 (d,J= 7.6 Hz, 2H), 7.61 (d,J= 7.6 Hz, 1H), 7.55~7.46 (m, 3H).13C NMR (100 MHz, CDCl3),δ: 171.29, 165.13, 148.60,140.58, 130.66, 129.76, 129.54, 129.20, 127.40, 126.82,124.15, 112.93.HRMS (EI): C14H9NO2S(M+),计算值255.0354,测试值222.0352。
(5-苯基-2-噻唑基)-2-吡咯甲酮 (C3): 黄色固体,产率32%,m.p.155.8~156.6 ℃;1H NMR (400 MHz, CDCl3),δ:11.08 (s, 1H), 8.10 (s, 1H), 7.59 (t,J= 1.8 Hz, 1H), 7.58~7.57(m, 1H), 7.54~7.47 (m, 1H), 7.42~7.33 (m, 3H), 7.12~7.08 (m,1H), 6.34 (dt,J= 4.0, 2.4 Hz, 1H).13C NMR (100 MHz,CDCl3),δ: 145.71, 139.75, 130.57, 129.50, 129.38, 128.67,127.31, 127.17, 127.11, 126.55, 126.08, 111.78.HRMS (EI):C14H10N2OS (M+),计算值254.0514,测试值254.0516。
(5-苯基-2-噻唑基)-3-噻吩甲酮 (C4): 黄色固体,产率25%,m.p.119.5~120.3 ℃;1H NMR (400 MHz, CDCl3),δ:9.07 (dd,J= 3.0, 1.2 Hz, 1H), 8.14 (s, 1H), 7.89 (dd,J= 5.2,1.2 Hz, 1H), 7.61 (t,J= 1.4 Hz, 1H), 7.59 (t,J= 1.4 Hz, 1H),7.42~7.34 (m, 3H), 7.30 (dd,J= 5.2, 3.0 Hz, 1H).13C NMR(100 MHz, CDCl3),δ: 177.23, 166.58, 146.55, 140.30, 138.56,137.20, 130.61, 129.56, 129.38, 128.81, 127.25, 125.65.HRMS (EI): C14H9NOS2(M+),计算值271.0126,测试值271.0130。
(5-(2-呋喃基)-2-噻唑基)-2-噻吩甲酮 (D1): 黄色固体,产率46%,m.p.123.5~124.3 ℃;1H NMR (400 MHz,CDCl3),δ: 8.62 (dd,J= 4.0, 1.0 Hz, 1H), 8.17 (s, 1H), 7.79(dd,J= 4.8, 1.0 Hz, 1H), 7.54 (d,J= 1.8 Hz, 1H), 7.23 (dd,J= 4.8, 4.0 Hz, 1H), 6.77 (d,J= 3.4 Hz, 1H), 6.53 (dd,J= 3.4,1.8 Hz, 1H).13C NMR (100 MHz, CDCl3),δ: 175.69, 164.55,145.91, 143.91, 139.76, 139.49, 136.73, 136.21, 135.81,128.34, 112.39, 109.81.HRMS (EI): C12H7NO2S2(M+),计算值260.9915,测试值260.9918。
(5-(2-噻吩基)-2-噻唑基)-2-噻吩甲酮 (D2): 褐色固体,产率22%,m.p.132.6~133.3 ℃;1H NMR (400 MHz,CDCl3),δ: 8.55 (dd,J= 3.8, 1.2 Hz, 1H), 8.03 (s, 1H), 7.73(dd,J= 4.8, 1.2 Hz, 1H), 7.34 (dd,J= 5.0, 1.0 Hz, 1H), 7.31(dd,J= 3.6, 1.0 Hz, 1H), 7.17 (t,J= 4.8, 4.0 Hz, 1H), 7.05(dd,J= 5.0, 3.6 Hz, 1H).13C NMR (100 MHz, CDCl3),δ:177.73, 158.14, 140.22, 139.71, 136.72, 136.21, 132.44,131.12, 128.43, 128.32, 127.40, 127.38.HRMS (ESI):C12H7NOS3(M + H)+,计算值277.9763,测试值277.9773。
(5-(3-噻吩基)-2-噻唑基)-2-噻吩甲酮 (D3): 黄色固体,产率25%,m.p.152.6~153.3 ℃;1H NMR (400 MHz,CDCl3),δ: 8.63 (dd,J= 3.8, 1.2 Hz, 1H), 8.13 (s, 1H), 7.79(dd,J= 5.0, 1.2 Hz, 1H), 7.61 (dd,J= 3.0, 1.4 Hz, 1H), 7.45(dd,J= 5.0, 3.0 Hz, 1H), 7.38 (dd,J= 5.0, 1.4 Hz, 1H), 7.23(dd,J= 5.0, 3.8 Hz, 1H).13C NMR (100 MHz, CDCl3),δ:175.75, 164.62, 141.29, 140.32, 139.77, 136.72, 136.19,131.33, 128.32, 127.49, 126.29, 123.63.HRMS (EI):C12H7NOS3(M+),计算值276.9690,测试值276.9688。
(5-([1,1 '-联苯基]-4-基)-2-噻唑基)-2-噻吩甲酮 (D4):黄色固体,产率35%,m.p.162.3~163.1 ℃;1H NMR (400 MHz, CDCl3),δ: 8.57 (dd,J= 4.0, 1.2 Hz, 1H), 8.19 (s, 1H),7.73 (dd,J= 5.0, 1.2 Hz, 1H), 7.69 (t,J= 1.8 Hz, 1H), 7.66 (t,J= 2.2 Hz, 1H), 7.63 (t,J= 2.2 Hz, 1H), 7.61 (t,J= 1.8 Hz,1H), 7.57 (t,J= 1.6 Hz, 1H), 7.56~7.54 (m, 1H), 7.44~7.37(m, 2H), 7.35~7.29 (m, 1H), 7.17 (dd,J= 4.9, 3.9 Hz, 1H).13C NMR (100 MHz, CDCl3),δ: 175.81, 165.51, 146.49, 142.44,140.26, 139.93, 139.75, 136.79, 136.26, 129.45, 129.01,128.35, 128.00, 127.98, 127.65, 127.04.HRMS (ESI):C20H13NOS2(M + H)+,计算值348.0511,测试值348.0507。
(5-(4-苯氧基苯酚)-2-噻唑基)-2-噻吩甲酮 (D5): 黄色固体,产率38%,m.p.150.5~151.2 ℃;1H NMR (400 MHz,CDCl3),δ: 8.63 (dd,J= 3.8, 1.0 Hz, 1H), 8.15 (s, 1H), 7.79(dd,J= 4.8, 1.0 Hz, 1H), 7.63 (t,J= 2.0 Hz, 1H), 7.61 (t,J=2.0 Hz, 1H), 7.39 (td,J= 7.6, 2.0 Hz, 2H), 7.23 (dd,J= 4.9,4.0 Hz, 1H), 7.18 (t,J= 7.4 Hz, 1H), 7.11~7.03 (m, 4H).13C NMR (100 MHz, CDCl3),δ: 175.79, 165.10, 158.93, 156.15,146.39, 139.80, 139.75, 136.71, 136.19, 130.02, 128.79,128.32, 125.25, 124.20, 119.63, 118.99.HRMS (ESI):C20H13N O2S2(M + H)+,计算值3 6 4.0 4 6 0,测试值:364.0454。
(5-(4-对甲苯硫基)苯基-2-噻唑基)-2-噻吩甲酮 (D6): 褐色固体,产率42%,m.p.163.4~164.3 ℃;1H NMR (400 MHz, CDCl3),δ: 8.62 (dd,J= 3.8, 1.2 Hz, 1H), 8.17 (s, 1H),7.79 (dd,J= 4.8, 1.2 Hz, 1H), 7.54 (d,J= 2.0 Hz, 1H), 7.52(d,J= 2.0 Hz, 1H), 7.41 (d,J= 1.8 Hz, 1H), 7.39 (t,J= 1.8 Hz, 1H), 7.25~7.19 (m, 5H), 2.39 (s, 3H).13C NMR (100 MHz,CDCl3),δ: 175.75, 165.26, 146.30, 140.76, 140.06, 139.71,138.85, 136.74, 136.24, 133.72, 130.45, 129.15, 128.82,128.32, 127.93, 127.60, 21.29.HRMS (ESI): C21H15NOS3(M + H)+,计算值394.0388,测试值394.0383。
(5-(3-甲基噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E1):黄色固体,产率51%,m.p.142.6~143.3 ℃;1H NMR (400 MHz, DMSO-d6),δ: 8.59 (dd,J= 4.0, 1.0 Hz, 1H), 8.35 (s,1H), 8.23 (dd,J= 4.8,1.0 Hz,1H), 7.69 (d,J= 5.0 Hz, 1H),7.38 (t,J= 4.4 Hz, 1H), 7.12 (d,J= 5.0 Hz, 1H), 3.35 (s, 3H).13C NMR (100 MHz, CDCl3),δ: 175.82, 165.15, 141.62,139.82, 139.38, 137.51, 136.70, 136.19, 131.94, 128.32,126.80, 125.96, 15.84.HRMS (ESI): C13H9NOS3(M + H)+,计算值291.9919,测试值291.9929。
(5-(4-甲基噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E2):褐色固体,产率42%,m.p.132.5~133.1 ℃;1H NMR (400 MHz, DMSO-d6),δ: 8.57 (dd,J= 4.0, 1.2 Hz, 1H), 8.41 (s,1H), 8.23 (dd,J= 4.8, 1.2 Hz,1H), 7.50 (s, 1H), 7.40~7.34 (m,2H), 3.35 (s, 3H).13C NMR (100 MHz, DMSO-d6),δ: 175.21,163.98, 141.23, 140.41, 139.48, 139.32, 138.39, 137.36,131.56, 130.87, 129.39, 124.66, 15.76.HRMS (ESI):C13H9NOS3(M + H)+,计算值291.9919,测试值291.9929。
(5-(5-甲基噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E3):褐色固体,产率43%,m.p.141.6~142.3 ℃;1H NMR (400 MHz, CDCl3),δ: 8.64 (d,J= 3.6 Hz, 1H), 8.04 (s, 1H), 7.82(d,J= 4.4 Hz, 1H), 7.28~7.23 (m, 1H), 7.20 (d,J= 2.2 Hz,1H), 6.79 (d,J= 2.2 Hz, 1H), 2.56 (s, 3H).13C NMR (100 MHz, DMSO-d6),δ: 175.18, 163.58, 143.45, 140.84, 140.52,139.34, 138.33, 137.29, 129.57, 129.37, 129.04, 127.91, 15.56.HRMS (ESI): C13H9NOS3(M + H)+,计算值291.9919,测试值291.9916。
(5-(3-溴噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E4):黄色固体,产率38%,m.p.135.1~135.6 ℃;1H NMR (400 MHz,DMSO-d6),δ: 8.61 (dd,J= 3.8, 1.0 Hz, 1H), 8.58 (s, 1H), 8.26(dd,J= 5.0, 1.0 Hz, 1H), 7.91 (d,J= 5.4 Hz, 1H), 7.41~7.38(m, 1H), 7.35 (d,J= 5.4 Hz, 1H).13C NMR (100 MHz,DMSO-d6),δ: 175.57, 165.40, 144.03, 142.47, 139.31, 138.67,137.60, 136.79, 132.77, 130.07, 129.51, 112.22.HRMS (ESI):C12H679BrNOS3(M + H)+,计算值355.8868,测试值355.8873; HRMS (ESI): C12H681BrNOS3(M + H)+,计算值357.8847,测试值357.8858。
(5-(4-溴噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E5):黄色固体,产率33%,m.p.145.8~146.4 ℃;1H NMR (400 MHz,DMSO-d6),δ: 8.58 (d,J= 3.8 Hz, 1H), 8.54 (s, 1H), 8.24 (d,J= 4.8 Hz, 1H), 7.90 (d,J= 1.2 Hz, 1H), 7.78 (d,J= 1.2 Hz,1H), 7.38 (t,J= 4.4 Hz, 1H).13C NMR (100 MHz, DMSO-d6),δ: 175.23, 165.13, 142.36, 139.19, 138.67, 138.51, 137.57,133.63, 130.45, 129.49, 126.61, 110.85.HRMS (ESI):C12H679BrNOS3(M + H)+,计算值355.8868,测试值355.8876; HRMS (ESI) C12H681BrNOS3(M + H)+,计算值357.8847,测试值357.8861。
(5-(5-溴噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E6):黄色固体,产率37%,m.p.143.1~143.9 ℃;1H NMR (400 MHz,CDCl3),δ: 8.54 (dd,J= 3.8, 1.2 Hz, 1H), 7.95 (s, 1H), 7.73(dd,J= 4.8, 1.2 Hz, 1H), 7.16 (t,J= 4.8, 3.8 Hz, 1H), 7.04 (d,J= 3.8 Hz, 1H), 6.99 (d,J= 3.8 Hz, 1H).13C NMR (100 MHz,DMSO-d6),δ: 175.21, 164.75, 141.99, 139.34, 138.76, 138.61,137.51, 132.69, 129.67, 129.47, 126.24, 114.55.HRMS (ESI):C12H679BrNOS3(M + H)+,计算值355.8868,测试值355.8874; HRMS (ESI): C12H681BrNOS3(M + H)+,计算值357.8847,测试值357.8849。
(5-(3-氯噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E7):黄色固体,产率34%,m.p.136.5~137.2 ℃;1H NMR (400 MHz,DMSO-d6),δ: 8.59 (dd,J= 3.6, 0.8 Hz, 1H), 8.54 (s, 1H), 8.23(dd,J= 4.8, 0.8 Hz, 1H), 7.90 (d,J= 5.4 Hz, 1H), 7.37 (t,J=4.4 Hz, 1H), 7.30 (d,J= 5.4 Hz, 1H).13C NMR (100 MHz,DMSO-d6),δ: 175.52, 165.49, 143.63, 139.30, 138.65, 137.57,136.06, 130.02, 129.49, 129.26, 126.17, 124.76.HRMS (ESI):C12H635ClNOS3(M + H)+,计算值311.9373,测试值311.9383; HRMS (ESI): C12H637ClNOS3(M + H)+,计算值313.9343,测试值313.9349。
(5-(4-氯噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E8):黄色固体,产率33%,m.p.127.8~128.4 ℃;1H NMR (400 MHz,CDCl3),δ: 8.65 (d,J= 3.0 Hz, 1H), 8.11 (s, 1H), 7.83 (d,J=4.2 Hz, 1H), 7.28~7.23 (m, 2H), 7.20 (d,J= 1.0 Hz, 1H).13C NMR (100 MHz, CDCl3),δ: 175.50, 165.70, 140.66, 139.59,138.59, 136.97, 136.53, 132.96, 128.45, 127.12, 126.70,121.48.HRMS (ESI): C12H635ClNOS3(M + H)+,计算值311.9373,测试值311.9383; HRMS (ESI): C12H637ClNOS3(M + H)+,计算值313.9343,测试值313.9351。
(5-(5-氯噻吩-2-基)-2-噻唑基)-2-噻吩甲酮 (E9):褐色固体,产率31%,m.p.134.3~134.9 ℃;1H NMR (400 MHz,DMSO-d6),δ: 8.54 (d,J= 3.8 Hz, 1H), 8.43 (s, 1H), 8.20 (d,J= 4.8 Hz, 1H), 7.54 (d,J= 4.0 Hz, 1H), 7.34 (t,J= 4.4 Hz,1H), 7.25 (d,J= 4.0 Hz, 1H).13C NMR (100 MHz, CDCl3),δ:175.51, 165.13, 140.22, 139.65, 139.00, 136.87, 136.41,132.16, 131.07, 128.41, 127.57, 126.66.HRMS (ESI):C12H635ClNOS3(M + H)+,计算值311.9373,测试值311.9384; HRMS (ESI): C12H637ClNOS3(M + H)+,计算值313.9343,测试值313.9352。
2.2 目标化合物对南方根结线虫的生物活性
为了进一步判断目标化合物对南方根结线虫是否具有活性,分别进行了离体和活体方面的测试,同时便于分析活性的原因。离体测试是为了判断化合物是否直接作用于线虫,导致其死亡。活体测试包括在沙土中和基质土壤中的测试。在沙土中测试,考察化合物在活体条件下对南方根结线虫造成的根结的抑制率,从而间接判断对线虫的抑制活性,只有沙子和植物以及营养液,影响因素少,便于分析化合物的构效关系,沙土中有活性可能是对线虫卵孵化的影响,或化合物影响了线虫的其他生理活动,如抑制线虫的摄食能力和运动能力等,使得线虫不再侵染根系。基质土壤测试则模拟了实际应用中田间的环境,加入了土壤pH 值和微生物对化合物的影响,有利于评价化合物是否具有使用价值。
2.2.1 化合物B1~B9、C1~C4、D1~D6 对南方根结线虫的生物活性 离体和活体测试结果如表1所示。第1 轮优化合成了化合物B2~B9,考察了不同芳环替换呋喃环后化合物活性的变化。结果表明,所合成的8 个化合物在40 mg/L 下72 h 时对南方根结线虫几乎没有离体致死活性;在活体沙土测试中,在40 mg/L 下含5-苯基噻吩的化合物B2 和含5-苯基噻唑的化合物B5 对南方根结线虫的抑制率分别为90.58%和100%,当质量浓度降低到5 mg/L 时,化合物B2 和B5 的抑制率依然超过了75%,其中化合物B5 的抑制率最高,达81.84%,且明显高于先导化合物B1。第2 轮优化合成了化合物C1~C4。测定结果表明,离体条件下,C2 在40 mg/L 下对南方根结线虫的致死率达到39.20%;在活体沙土中,含3-噻吩的化合物C4在40 mg/L 下对南方根结线虫的抑制率为96.27%,但降低质量浓度后抑制率直接降为0。由此可见,依旧是含2-噻吩的化合物B5 的活性最好。第3 轮优化合成了化合物D1~D6。测定结果表明,离体条件下,化合物D1 在40 mg/L 下72 h 时对南方根结线虫的致死率为70.03%,D2 的致死率为42.29%。但在沙土中,40 mg/L 的化合物D1 基本丧失了活性,而化合物D2 的抑制率为99.43%,且在5 mg/L 下化合物D2 仍有87.61%的抑制率,略高于化合物B5,明显高于先导化合物B1,但低于阳性对照噻唑膦。基于上述结果,经综合比较后,确定D2 为活性最高的骨架化合物,其结构为 (5-(2-噻吩基)-2-噻唑基)-2-噻吩甲酮,在此基础上进行了第4 轮优化,合成了化合物E1~E9。

表1 化合物B1~B9、C1~C4、D1~D6 在离体和活体条件下对南方根结线虫的活性Table 1 In vitro and in vivo nematicidal activity of compounds B1-B9、C1-C4、D1-D6 against M. incognita
2.2.2 化合物E1~E9 对南方根结线虫的生物活性
测试结果(表2)表明:将取代基引入化合物D2的噻吩环上,只有化合物E3 (R = 5-CH3) 在40 mg/L下72 h 时对南方根结线虫有71.26% 的致死率。在沙土测试中,化合物E3 丧失了活性;化合物E6 (R = 5-Br) 和E9 (R = 5-Cl) 在40 mg/L 下对南方根结线虫造成的根结抑制率分别为91.49%和95.16%,两者活性与D2 基本相当;相同质量浓度下,化合物E2 (R = 4-CH3)、E3 (R = 5-CH3)、E5 (R = 4-Br) 和E8 (R = 4-Cl) 的抑制率也分别达到76.33%、53.31%、65.38%和85.52%。初步的构效关系分析表明,当噻吩环上的取代基为吸电子基时 (E5 (R = 4-Br)、E6 (R = 5-Br)、E8 (R = 4-Cl) 和E9 (R = 5-Cl) ) 的活性优于取代基为给电子基 (E2 (R = 4-CH3) 和E3 (R = 5-CH3) ) 的活性,且5-位 > 4-位 > 3-位,但当质量浓度降到5 mg/L时,E6 (R = 5-Br) 和E9 (R = 5-Cl) 的抑制率分别为45.04%和75.35%,均明显低于D2。

表2 化合物E1~E9 在离体及活体条件下对南方根结线虫的活性Table 2 In vitro and in vivo nematicidal activity of compounds E1-E9 against M. incognita
2.2.3 化合物B2、B5、D2、E8 和E9 在不同土壤 (基质) 中对南方根结线虫的生物活性 考虑到化合物B2、B5、D2、E8 和E9 在低质量浓度(5 mg/L)沙土中表现出较好的对由南方根结线虫造成的根结抑制活性,进一步测试了这5 种化合物在基质土壤中对植物根结的抑制活性。结果发现,化合物B2、B5、D2 和E9 在40 mg/L 质量浓度下的根结抑制率均在55%以上,其中B5 的抑制率最高,达77.67%,优于化合物A,也优于B1,但还低于噻唑膦。化合物B2 和E9 在40 mg/L 下对植物根结的抑制率分别为75.90% 和64.30%,高于B1 和D2,但低于B5。
分析3 种测试结果,可以看出,只有少数几个化合物C2、D1、D2 和E3 在离体条件下对南方根结线虫具有一定的致死率,推测可能是其作用于线虫神经系统等导致线虫直接死亡,但直接致死作用不强。在沙土中,化合物B2、B5、E1、E2、E6、E8 和E9 对由南方根结线虫造成的植物根结的抑制活性明显,但这些化合物在离体条件下基本没有活性,说明化合物可能主要影响了线虫的其他生理活动,如抑制卵孵化、摄食能力和运动能力等[26],使得线虫不易侵染植物根系;也有可能是化合物诱导了植物产生系统性抗性,使得线虫不易侵染植物根系[36-37];化合物D2 不仅对南方根结线虫具有中等的离体致死作用,而且也可以影响线虫侵染植物根系,最终可能是对线虫的直接致死作用和抑制线虫其他生理活动的协同作用的结果。在基质土壤中,化合物B2、B5、D2和E9表现出较好对植物根结的抑制率,其中B5活性最高。以上结果表明双杂环基甲酮结构并不是直接作用于南方根结线虫而产生生物活性,可能对线虫的卵孵化和其他线虫生理活动造成影响或者诱导植物产生系统性抗性,深入的作用机制还待进一步研究。
3 结论
本文基于稠环优化策略,将苯并呋喃变换为苯连呋喃,设计合成了27 个未见文献报道的双杂环基甲酮类化合物,并对这些化合物进行了杀线虫活性的测定。结果表明:除化合物D1和E3在40 mg/L 下72 h 时对南方根结线虫的致死率分别为70.03%和71.26%外,其余大部分化合物对其没有直接的致死活性;在40 mg/L 时化合物B1、B2、B5、C4、D2、E6和E9在沙土中对由南方根结线虫造成的植物根结的抑制率均在90% 以上,其中D2在5 mg/L 时抑制率最高,为87.61%,且高于先导化合物B1;而在基质土壤中的测试结果表明,化合物B5的抑制活性最高,在40 mg/L时抑制率为77.67%,化合物D2的活性低于B5,但均高于先导化合物B1,说明将苯并呋喃变换为苯连呋喃的设计策略是成功的。初步的构效关系研究表明,不含任何取代基的(5-(2-噻吩基)-2-噻唑基)-2-噻吩甲酮化合物具有更好的杀线虫活性,这为进一步的分子设计提供了思路。
猜你喜欢
云南化工(2021年7期)2021-12-21
益寿宝典(2017年1期)2017-09-03
中小学实验与装备(2016年1期)2016-04-19
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
郑州大学学报(工学版)(2015年1期)2015-03-24
红领巾·探索(2014年8期)2014-10-10
应用化工(2014年9期)2014-08-10
中国兽药杂志(2012年4期)2012-11-06
化学与生物工程(2012年9期)2012-07-28
