
HPAM/PPG颗粒悬浮液驱油体系增黏机制的分子模拟
2023-06-20 01:55:28姜祖明元福卿郝喜顺燕友果
中国石油大学学报(自然科学版) 2023年3期
姜祖明, 石 静, 元福卿, 祁 凯, 郝喜顺, 李 振, 燕友果
(1.中国石油化工股份有限公司胜利油田分公司勘探开发研究院,山东东营 257015;2.中国石油大学(华东)材料科学与工程学院,山东青岛 266580)
聚合物驱在室内和矿场上得到全面的试验,已成为油田开发中改善水驱和提高采收率最为成熟的技术之一[1-2]。聚合物驱后油藏仍有大量剩余油,但由于储层非均质性更严重且剩余油更分散,开采难度大。悬浮液化学驱为聚合物驱后油藏提高采收率的新方法,它通过在聚合物HPAM(聚丙烯酰胺)溶液中引入可暂堵及变形运移的“软固体”黏弹性颗粒(PPG(聚丙二醇)颗粒),形成的聚合物HPAM/PPG颗粒悬浮液体系强化调驱效力,提高波及系数和洗油效率,进而提高原油采收率[3-4]。聚合物HPAM/PPG颗粒悬浮液体系的黏度是决定驱油效果的一个重要参数。悬浮液体系中溶解的聚合物HPAM通过形成空间网络结构实现结构增黏,同时聚合物HPAM表面的亲水基团形成水化层束缚周围的水分子,实现水动力学增黏。黏弹性PPG颗粒具有“部分交联部分支化”分子结构,其表面的线性支化链与聚合物HPAM分子链以及溶剂水之间的相互作用对悬浮液体系的黏度产生复杂的影响。可以通过流变试验测量不同质量浓度配比下的悬浮液体系黏度[5-6]。笔者采用分子动力学模拟方法,通过构建聚合物HPAM/PPG悬浮液模型,选取合适的分子力场及相互作用参数,从分子原子层次考察聚合物与PPG颗粒间的相互作用,揭示聚合物HPAM/PPG颗粒悬浮液体系的增黏机制。
1 模型和方法
HPAM的相对分子质量往往在几十万甚至上百万以上[7-10],由于聚丙烯酰胺是由重复单元构成的,因此选取聚丙烯酰胺的部分片段,采用粗粒度分子动力学(CGMD)模拟研究部分水解聚丙烯酰胺和预交联凝胶颗粒不同质量浓度比复合体系的黏度。模拟采用的HPAM的聚合度为80,水解度为40%。利用Materials Studio 软件构建HPAM部分片段全原子模型如图1(a)所示。根据MARTINI力场[11-15]粗粒化的映射规则(图1(b)),将全原子结构映射到HPAM的粗粒化模型中,如图1(c)所示。PPG颗粒的构建是通过将6条HPAM链段预交联形成核心获得,PPG颗粒的分子构型如图1(d)所示。图1(b)中各粗粒度珠子键相互作用参数见表1。其中成键包括两个类型:P5-SC1和Qa-SC1,键的相互作用参数为Rbond和Kbond;键角包括SC1-P5-SC1和P5-SC1-P5,键角的相互作用参数为θ0和Kangle。

表1 各粗粒度珠子键相互作用参数Table 1 Bond interaction parameters of coarse-grained beads

图1 试验模型Fig.1 Experimental models
为方便对比,控制聚合物HPAM/PPG颗粒复合调驱体系的总质量浓度为17 g/L,通过改变聚合物HPAM和PPG颗粒的质量浓度比,分别构建PPG颗粒质量浓度占比的5种复合体系。模拟体系中加入89640个极化水珠子,并通过调节Na+的数量,使复合体系保持电中性。采用Gromacs 4.5.5软件包[16]进行分子动力学模拟计算,采用VMD软件[17]用于模拟结果的可视化。模拟在X、Y、Z三个方向均采用周期性边界条件。模拟体系尺寸为30 mm×30 mm×30 nm。模拟采用NPT系综,通过Berendsen方法[18]控温控压,温度设为300 K,压力为0.1 MPa。首先采用最速下降法优化初始构型,对体系能量最小化,然后进行10 ns 的分子动力学模拟。采用PME粒子网格Ewald求和方法计算静电相互作用,模拟步长设定为1 fs。
2 结果分析
2.1 黏度变化规律
图2给出不同质量浓度比下聚合物HPAM/PPG颗粒复合体系平衡后的构型图以及模拟计算的黏度。为清晰显示聚合物HPAM和PPG颗粒的空间分布形态,隐藏了体系中的水。利用LAMMPS软件中的Muller-Plathe反向非平衡分子动力学方法[19]进行体系黏度计算,该方法的原理是动量在模拟盒子的2个不同层中的原子之间交换,从而在体系中产生剪切速度,根据剪切速度曲线获得体系中粒子的动量,基于线性拟合求解剪切速度曲线的斜率,进而计算体系的剪切黏度。黏度计算结果表明,随着PPG颗粒质量浓度增加,聚合物HPAM/PPG颗粒复合体系的黏度呈现出单调下降的趋势。黏度的变化表明,在相同的质量浓度下,舒展的聚合物HPAM具有更大的黏度,而交联形成聚集状态的PPG颗粒则具有较低的黏度。黏度是影响聚合物HPAM/PPG颗粒复合体系调驱性能的重要物性参数,为了揭示复合体系中聚合物HPAM和PPG颗粒对黏度的贡献,本文中从复合体系空间结构分布、水动力学以及能量角度剖析复合体系中黏度变化的分子机制。
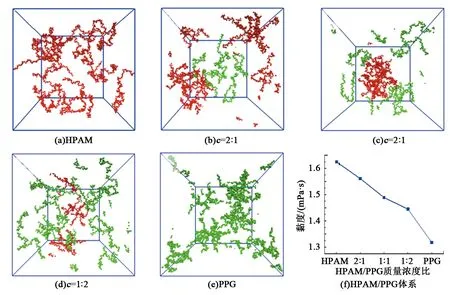
图2 不同质量浓度比下聚合物HPAM/PPG颗粒复合体系的平衡构型Fig.2 Equivalent configuration of polymer HPAM/PPG particles suspension flooding system
2.2 黏度变化的微观机制
2.2.1 结构增黏
研究表明,聚合物溶液的结构黏度在很大程度上决定于聚合物空间展布的有效长度[20-22]。聚合物链有效长度越大,越有利于其在空间相互搭接形成网络结构,网络结构捕获水抑制溶液的运动,从而获得较高的结构黏度。与聚合物HPAM不同,PPG颗粒通常是通过交联形成的,交联结构占据较大质量,只有暴露在外面的链才对黏度有贡献。为了定量描述聚合物HPAM和PPG颗粒在溶液中的展布状态,确定聚合物HPAM和PPG颗粒对黏度的贡献,这里计算了聚合物HPAM分子和PPG颗粒的回转半径Rg。回转半径的计算是采用最小的一个球面将考察的分子包裹在内,这个球的半径称为分子链的回转半径。通过回转半径可以表征分子的弯曲和伸展程度,回转半径大说明分子链舒展,对空间结构黏度的贡献大;反之说明分子链团聚,对空间黏度的贡献小。通过对模拟体系平衡后的所有聚合物HPAM(或PPG颗粒)统计平均,得到的聚合物HPAM和PPG颗粒的回转半径Rg如图3所示。

图3 聚合物HPAM和PPG颗粒的回转半径Fig.3 Gyration radius of polymer HPAM and PPG particle during simulation
图3表明,溶液中聚合物HPAM分子的回转半径大于PPG颗粒的回转半径。这一结果说明相对于PPG颗粒,聚合物HPAM分子链更容易搭接形成空间网络结构促进体系增黏。因此,在总质量浓度相同的情况下,当聚合物HPAM质量浓度占比增加时,可以形成更多的搭接点,从而有利于体系黏度的增大。这一分析结果可以很好地验证图2中黏度计算的结果,即当聚合物HPAM/PPG颗粒溶液体系中聚合物HPAM占比高时,体系具有更高的黏度。
在分子热运动的作用下,聚合物间缠结点处于不断的解体和重建的动态平衡中,具有瞬变的空间网状结构,聚合物溶液的黏度与聚合物间的缠结点数目成正比[23-24]。通过计算聚合物HPAM和PPG颗粒在模拟过程中形成的缠结点数目,选择整体溶液质量浓度为17 g/L,其中聚合物HPAM与PPG颗粒的质量浓度比(w)为0、0.5、1.0的3种体系进行研究,结果如图4(a)所示。可以看出,在相同质量浓度下,纯的聚合物HPAM因为具有更长的有效分子链长,因此在模拟中形成缠结点的数量最多,更可能形成交联网络实现结构增黏;而PPG颗粒由于分子链段较短,不容易实现交联形成空间网络结构,导致黏度较低;聚合物HPAM与PPG颗粒的混合溶液形成缠结点的能力居于两者之间。为了进一步证实以上结果,本文将溶液的质量浓度提高1倍进行考察,图4(b)给出不同质量浓度的对比结果。可以看出,当质量浓度提高后,聚合物HPAM和PPG颗粒体系分子链的交联数目有明显的提高。这一结果进一步表明聚合物HPAM和PPG颗粒含量高的交联网络结构可以有效实现增黏。

图4 三类体系交联数累积及不同质量浓度下3种交联类型交联数累积Fig.4 Accumulated cross-linking number of three systems and three cross-linking types at different mass concentrations
2.2.2 水动力学增黏
水溶液体系中聚合物HPAM分子和PPG颗粒含有酰胺—CONH2亲水基团以及水解后羧酸根—COO-两类亲水基团。这些亲水基团中的杂原子通过氢键和静电作用可以将极性水分子束缚在附近,形成一层排列规整、有序而紧密的水化层;同时水化层内的水分子彼此通过氢键作用加强了水化层的稳定性[25-26]。水化层的存在增加了聚合物HPAM和PPG颗粒链段的水动力学半径,导致分子链的运动受到抑制,在一定程度上提高了溶液黏度。因此束缚水数量越多,束缚强度越高,体系黏度越大。这里选取PPG颗粒质量浓度比为0.5的模拟体系作为典型代表进行研究,分别以—CONH2和—COO-基团为中心分子,计算聚合物HPAM分子和PPG颗粒上的两类亲水基团与水分子间的径向分布函数g(r),结果如图5所示。
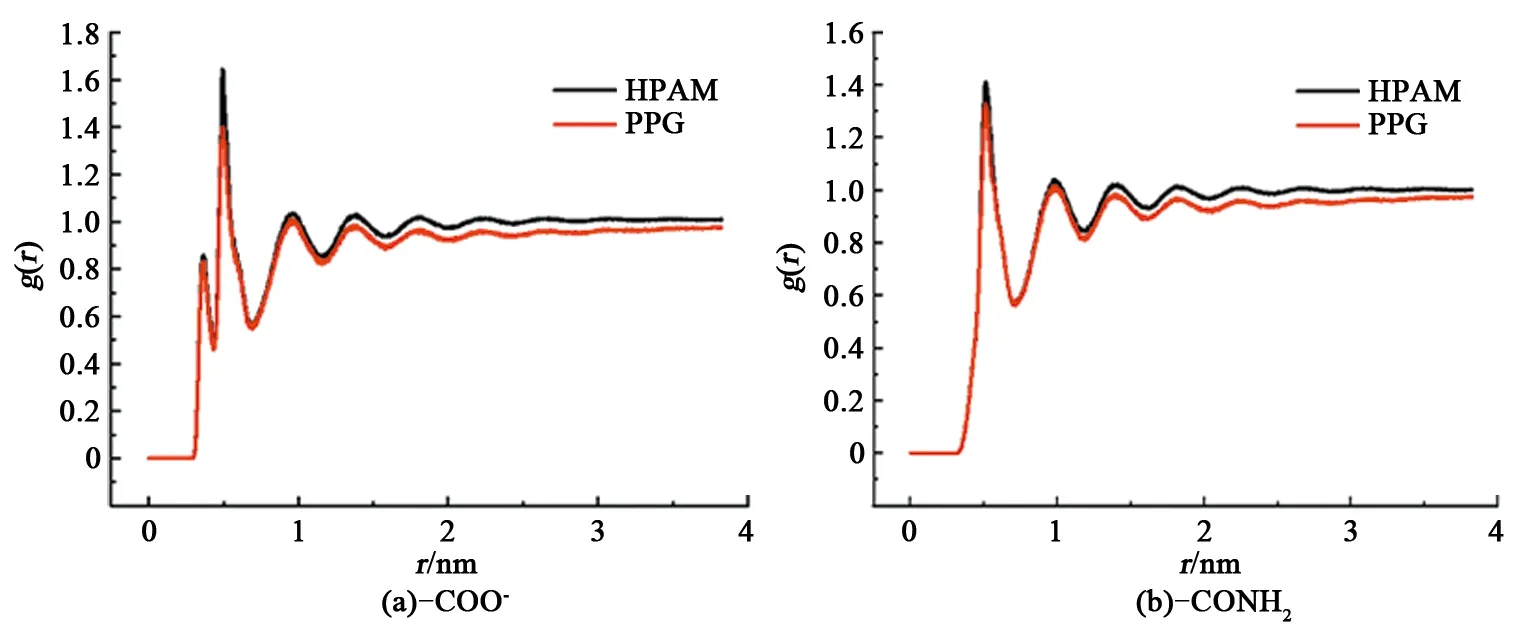
图5 聚合物HPAM和PPG颗粒中亲水基团对水分子径向分布函数Fig.5 Radial distribution function of hydrophilic group of polymer HPAM and PPG particle to water molecules
从图5看出,聚合物HPAM和PPG颗粒中的—CONH2和—COO-基团均与水分子形成了水化层。对于—CONH2基团,2种聚合物分子第一水化层的半径均约为6 Å,第一水化层的峰值大于1,意味着第一水化层水分子的密度要大于体相水的密度。对于—COO-基团,第一水化层的半径约为4.5 Å,峰值较小,这是由于—COO-特殊的空间位阻导致的,只有少量水分子进入到—COO-的内部位置;第二水化层约为6 Å,与—CONH2基团的第一水化层相当。
对比两类亲水基团对黏度的贡献表明,—COO-比—CONH2对水的束缚能力更强。这一点可以从亲水基团结合水的数量和结合水的强度得到证实。首先从径向分布函数分析结合水的数量,相比—CONH2,—COO-在6 Å附近具有更高的峰值;在4.5 Å附近—COO-还有一个额外的峰。这表明—COO-可以束缚更多的水分子,对黏度的贡献更大。同时还考察这两类基团在聚合物HPAM和PPG颗粒中对水分子束缚能力的差异。图5表明,两类基团在聚合物HPAM中具有更高的峰值,说明可以束缚更多的水分子,具有更高的黏度贡献。这一结果可以归咎于聚合物HPAM舒展和PPG颗粒卷曲的分子链结构。水分子在接触舒展的聚合物HPAM时空间位阻更小,聚合物HPAM亲水基团结合水的数量更多;与卷曲的PPG颗粒接触时由于空间位阻较大,PPG颗粒亲水基团结合水的数量就少。
进一步考察亲水基团—CONH2和—COO-对水分子的束缚能力。通过引入均力势(potential of mean force,PMF)来表征亲水基团与水分子作用的强弱,计算结果如图6所示。外部自由的水分子靠近亲水基团被捕获成为水化层中束缚水,在这一过程中所要克服的能垒即为溶剂化能ΔE+,如图6(a)所示;解离能ΔE-则表示水化层内的水分子脱离水化层成为自由水分子所需要克服的能垒,如图6(b)所示。为解释溶剂化能和解离能垒的计算过程,这里以—CONH2基团为例进行说明:①势能曲线极小值点(CM)约在0.52 nm处,是第一水化层中水分子与亲水基团的距离,其势能表明第一水化层中水分子与亲水基团的结合强度ECM;②势能曲线上的第二极小值点约在0.91 nm处,是溶剂分离极小值点(SSM),为第二水化层中水分子与亲水基团的距离,其数值表明第二水化层中水分子与亲水基团的结合强度ESSM;③CM和SSM中间存在一个比较高的能垒,称为溶剂层能垒(EBS),表示水分子从第二水化层进入第一水化层,需要克服的能垒。图6表明,溶液中的水分子进入第一水化层与亲水基团结合时,需要克服的最大能垒(即溶剂化能)是从SSM跨越过BS,ΔE+=EBS-ESSM。第一水化层的水分子解离时,需要克服的最大能垒(即解离能)是从CM跨越过BS,ΔE-=EBS-ECM。
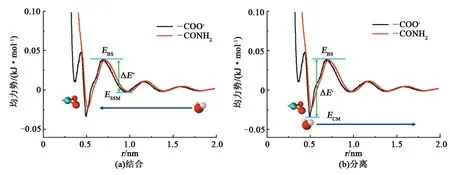
图6 亲水基团与水分子结合和分离时的均力势曲线Fig.6 Potential mean force profile during combination and separation of water molecule to hydrophilic groups
表2为根据图6中各个能量数据计算得到的—COO-和—CONH2的溶剂化能和解离能数值。可以看出,形成水化层所需要克服的溶剂化能比水化层中水分子的解离能小,说明自由水分子进入水化层后不容易逃逸,进而表明这两类亲水基团均可以形成稳定的水化层;—COO-与—CONH2相比,在形成水化层时需要克服的溶剂化能更小,说明更容易结合水形成水化层;水分子从—COO-水化层解离时,与—CONH2相比,需要克服更大的解离能,说明对水化层中水的结合更为牢固。因此与—CONH2相比,—COO-更容易形成水化层,而且形成的水化层更稳固,对黏度具有更大的贡献。

表2 亲水基团与水分子间的溶剂化能ΔE+和解离能ΔE-Table 2 Solvation energy and dissociation energy between hydrophilic groups with water molecules
3 结 论
(1)对于聚合物HPAM/PPG颗粒悬浮液体系,在相同总的质量浓度下,聚合物HPAM的占比增加提供了更高的体系黏度。
(2)相比PPG颗粒,聚合物HPAM在悬浮液中具有更大的回转半径,其舒展的分子结构相互搭接,有利于形成空间网络结构,实现大的结构增黏;聚合物HPAM和PPG颗粒分子链段上的亲水基团—CONH2和—COO-与水分子具有较强的作用,可以形成稳定的水化层,束缚水分子实现水动力学增黏。
(3)相比—CONH2,—COO-水化层形成过程中所需克服的溶剂化能更低,因此周围水分子数量更多;同时水化层中水分子的解离能更大,表明结合强度更大。以上结果说明—COO-对水动力学黏度的贡献更大。此外聚合物HPAM的亲水基团比PPG颗粒的亲水基团对水动力学黏度的贡献更大。
猜你喜欢
中国计量大学学报(2023年3期)2023-11-01 08:33:46
科教新报(2021年11期)2021-05-12 19:50:11
理化检验-化学分册(2020年12期)2021-01-26 00:41:40
科学之谜(2016年9期)2016-10-11 08:59:04
中国测试(2016年11期)2016-04-01 03:53:04
体育世界(学术版)(2015年3期)2015-07-01 17:15:41
应用化工(2014年11期)2014-08-16 15:59:13
食品科学(2013年14期)2013-03-11 18:25:13
少儿科学周刊·儿童版(2012年5期)2012-08-30 20:12:50
中国重型装备(2010年1期)2010-11-29 11:30:10
